Freyja Workflow Series¶
Quick Facts¶
| Workflow Type | Applicable Kingdom | Last Known Changes | Command-line Compatibility | Workflow Level | Dockstore |
|---|---|---|---|---|---|
| Genomic Characterization | SARS-CoV-2, Viral | vX.X.X | Yes | Sample-level, Set-level | Freyja_FASTQ_PHB, Freyja_Plot_PHB, Freyja_Dashboard_PHB |
Freyja Overview¶
Freyja is a tool for analyzing viral mixed sample genomic sequencing data. Developed by Joshua Levy from the Andersen Lab, it performs two main steps:
- Variant Frequency Estimation: Freyja calculates the frequencies of single nucleotide variants (SNVs) in the genomic sequencing data.
- Depth-Weighted Demixing: It separates mixed populations of viral subtypes using a depth-weighted statistical approach, estimating the proportional abundance of each subtype in the sample based on the frequencies of subtype-defining variants.
Additional post-processing steps can produce visualizations of aggregated samples.
Wastewater and more
The typical use case of Freyja is to analyze mixed SARS-CoV-2 samples from a sequencing dataset, most often wastewater, but the tool is not limited to this context. With the appropriate reference genomes and barcode files, Freyja can be adapted for other pathogens, including MPXV, Influenza, RSV, and Measles.
Default Values
The defaults included in the Freyja workflows reflect this use case but can be adjusted for other pathogens. See the Running Freyja on other pathogens section for more information. Please be aware this is an experimental feature and we cannot guarantee complete functionality at this time.
Figure 1: Freyja Suite Workflow Overview
Figure 1¶
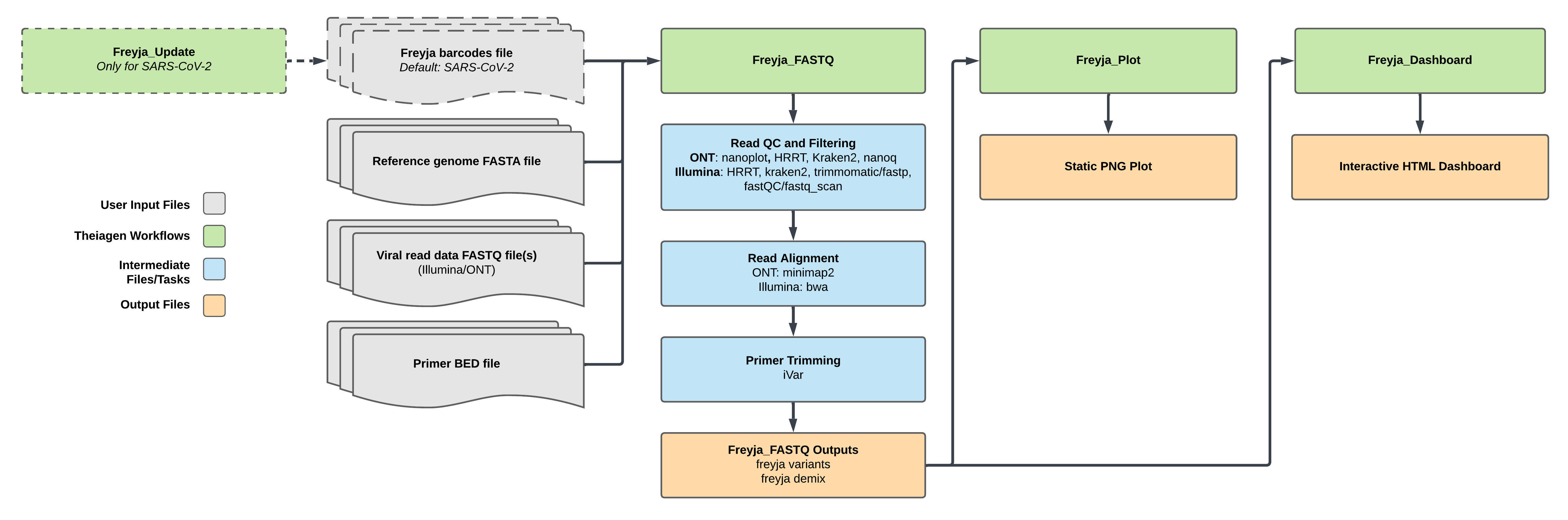
Depending on the type of data (Illumina or Oxford Nanopore), the Read QC and Filtering steps, as well as the Read Alignment steps use different software. The user can specify if the barcodes and lineages file should be updated with freyja update before running Freyja or if bootstrapping is to be performed with freyja boot.
Three workflows have been created that perform different parts of Freyja:
The main workflow is Freyja_FASTQ_PHB (Figure 1). Depending on the type of input data (Illumina paired-end, Illumina single-end or ONT), it runs various QC modules before aligning the sample with either BWA (Illumina) or minimap2 (ONT) to the provided reference file, followed by iVar for primer trimming. After the preprocessing is completed, Freyja is run to generate relative lineage abundances (demix) from the sample. Optional bootstrapping may be performed.
Data Compatibility
The Freyja_FASTQ_PHB workflow is compatible with the following input data types:
- Illumina Single-End
- Illumina Paired-End
- Oxford Nanopore
Two options are available to visualize the Freyja results: Freyja_Plot_PHB and Freyja_Dashboard_PHB. Freyja_Plot_PHB aggregates multiple samples using output from Freyja_FASTQ_PHB to generate a plot that shows fractional abundance estimates for all samples, including the option to plot sample collection date information. Alternatively, Freyja_Dashboard_PHB aggregates multiple samples using output from Freyja_FASTQ_PHB to generate an interactive visualization. This workflow requires an additional input field called viral load, which is the number of viral copies per liter.
Freyja, Sequencing Platforms and Data Quality¶
The choice of sequencing platform and the quality of the data directly influence Freyja's performance. High-accuracy platforms like Illumina provide reliable SNV detection, enhancing the precision of lineage abundance estimates. In contrast, platforms with higher error rates, such as ONT, may introduce uncertainties in variant calling, affecting the deconvolution process. Sequencing depth requirements will increase as the quality of the sequencing data decreases. A rational target depth is 100X coverage for sequencing data with Q-scores in the range of 25-30.
Additionally, inadequate sequencing depth can hinder Freyja's ability to differentiate between lineages, leading to potential misestimations. Sequencing depth requirements will increase with the complexity of the sample composition and the diversity of lineages present. For samples containing multiple closely related lineages, higher sequencing depth is necessary to resolve subtle differences in genetic variation and accurately estimate lineage abundances. This is particularly important for pathogens with high mutation rates or a large number of cocirculating lineages, such as influenza, where distinguishing between lineages relies on detecting specific single nucleotide variants (SNVs) with high confidence.
Freyja Workflows¶
Freyja_FASTQ_PHB¶
Freyja measures SNV frequency and sequencing depth at each position in the genome to return an estimate of the true lineage abundances in the sample. The method uses lineage-defining "barcodes" that, for SARS-CoV-2, are derived from the UShER global phylogenetic tree as a base set for demixing. Freyja_FASTQ_PHB returns as output a TSV file that includes the lineages present and their corresponding abundances, along with other values. Optionally, the workflow can also produce a long-format TSV (freyja_parsed_format_tsv) that pairs the demixed lineage abundances with sample metadata (collection date, collection site, latitude, longitude) for downstream visualization.
The Freyja_FASTQ_PHB workflow is compatible with the multiple input data types: Illumina Single-End, Illumina Paired-End and Oxford Nanopore. Depending on the type of input data, different input values are used.
Table 1: Freyja_FASTQ_PHB input configuration for different types of input data.
| Table Columns | Illumina Paired-End | Illumina Single-End | Oxford Nanopore |
|---|---|---|---|
| read1 | ✅ | ✅ | ✅ |
| read2 | ✅ | ❌ | ❌ |
| ont | false |
false |
true |
Inputs¶
This workflow runs on the sample level.
| Terra Task Name | Variable | Type | Description | Default Value | Terra Status |
|---|---|---|---|---|---|
| freyja_fastq | read1 | File | FASTQ file containing read1 sequences (Illumina or (ONT) | Required | |
| freyja_fastq | reference_genome | File | The reference genome to use; should match the reference used for alignment (Wuhan-Hu-1) | Required | |
| freyja_fastq | samplename | String | The name of the sample being analyzed | Required | |
| freyja_fastq | freyja_lineage_metadata | File | File containing the lineage metadata; the "curated_lineages.json" file found https://github.com/andersen-lab/Freyja/tree/main/freyja/data can be used for this variable. Does not need to be provided if update_db is true or if the freyja_pathogen is provided. | Optional, Required | |
| bwa | cpu | Int | Number of CPUs to allocate to the task | 6 | Optional |
| bwa | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| bwa | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| bwa | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| freyja | adapt | Float | adaptive lasso penalty parameter | 0.0 | Optional |
| freyja | auto_adapt | Boolean | When set to true will use error profile to set adapt value | False | Optional |
| freyja | bootstrap | Boolean | Perform bootstrapping | False | Optional |
| freyja | confirmed_only | Boolean | Include only confirmed SARS-CoV-2 lineages | False | Optional |
| freyja | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| freyja | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/freyja:2.0.1 | Optional |
| freyja | eps | Float | The minimum lineage abundance cut-off value | 0.001 | Optional |
| freyja | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| freyja | number_bootstraps | Int | The number of bootstraps to perform (only used if bootstrap = true) | 100 | Optional |
| freyja | update_db | Boolean | Updates the Freyja reference files (the usher barcodes and lineage metadata files) but will not save them as output (use Freyja_Update for that purpose). If set to true, the freyja_lineage_metadata and freyja_barcodes files are not required. | False | Optional |
| freyja_fastq | collection_date | String | Collection date of wastewater sample (YYYY-MM-DD) | Optional | |
| freyja_fastq | collection_site | String | Collection site of wastewater sample | Optional | |
| freyja_fastq | depth_cutoff | Int | The minimum coverage depth with which to exclude sites below this value and group identical barcodes -- THIS MAY NOT WORK FOR NON-SARS-COV-2 ORGANISMS! | Optional | |
| freyja_fastq | freyja_barcodes | File | Custom barcode file. Does not need to be provided if update_db is true if the freyja_pathogen is provided. | Optional | |
| freyja_fastq | freyja_long_format_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/freyja-microreact:1.0.2 | Optional |
| freyja_fastq | freyja_min_coverage | Int | Minimum genome coverage threshold (--mincov) used by freyja_to_long.py when generating the freyja_parsed_format_tsv output | 60 | Optional |
| freyja_fastq | freyja_pathogen | String | Pathogen to be used by Freyja | SARS-CoV-2 | Optional |
| freyja_fastq | kraken2_target_organism | String | The organism whose abundance the user wants to check in their reads. This should be a proper taxonomic name recognized by the Kraken database. | Severe acute respiratory syndrome coronavirus 2 | Optional |
| freyja_fastq | latitude | Float | Latitude of wastewater sample collection site | Optional | |
| freyja_fastq | longitude | Float | Longitude of wastewater sample collection site | Optional | |
| freyja_fastq | ont | Boolean | Indicates if the input data is derived from an ONT instrument. | False | Optional |
| freyja_fastq | primer_bed | File | The bed file containing the primers used when sequencing was performed | Optional | |
| freyja_fastq | qc_check_table | File | TSV containing values to check quality control metrics | Optional | |
| freyja_fastq | read2 | File | Illumina reverse read file in FASTQ file format (compression optional) | Optional | |
| freyja_fastq | reference_gff | File | The GFF file for reference; should match the reference used for alignment (Wuhan-Hu-1) | Optional | |
| freyja_fastq | run_qualimap | Boolean | When set to true, will run qualimap and provide custom visuals | True | Optional |
| freyja_fastq | sc2_gene_bed | File | BED file depicting SARS-CoV-2 region / gene coordinates | gs://theiagen-public-resources-rp/reference_data/viral/sars-cov-2/sc2_gene_locations.bed | Optional |
| freyja_fastq | trimmomatic_min_length | Int | The minimum length cut-off when performing read cleaning | 25 | Optional |
| freyja_long_format | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja_long_format | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| freyja_long_format | group_by | String | Whether to group samples by collection date or week, options are "date" or "week" | Optional | |
| freyja_long_format | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 4 | Optional |
| gene_coverage | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| gene_coverage | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| gene_coverage | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/pysam:1.23.1 | Optional |
| gene_coverage | exact_match | Boolean | Search for exact matches to query_genes / BED file gene queries | False | Optional |
| gene_coverage | feature_qualifier | String | Feature qualifier to use in parsing GBFF for coverage calculations | product | Optional |
| gene_coverage | feature_type | String | Feature type to use in parsing GBFF for coverage calculations | CDS | Optional |
| gene_coverage | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| gene_coverage | min_depth | Int | Minimum depth to call a base covered | 10 | Optional |
| gene_coverage | min_quality | Int | Minimum base quality to call a base covered | 0 | Optional |
| gene_coverage | query_genes | String | Comma-separated list of query genes to parse GBFF/BED file for region coordinates | Optional | |
| gene_coverage | reference_gbff | File | GenBank flat file depicting the reference genome | Optional | |
| get_fasta_genome_size | cpu | Int | Number of CPUs to allocate to the task | 1 | Optional |
| get_fasta_genome_size | disk_size | Int | Amount of storage (in GB) to allocate to the task | 10 | Optional |
| get_fasta_genome_size | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/seqkit:2.4.0--h9ee0642_0 | Optional |
| get_fasta_genome_size | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| minimap2 | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| minimap2 | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| minimap2 | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/minimap2:2.22 | Optional |
| minimap2 | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| minimap2 | query2 | File | Internal component, do not modify | Optional | |
| nanoplot_clean | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_clean | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_clean | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_clean | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_clean | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| nanoplot_raw | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_raw | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_raw | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_raw | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_raw | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| primer_trim | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| primer_trim | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| primer_trim | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| primer_trim | keep_noprimer_reads | Boolean | Include reads with no primers | True | Optional |
| primer_trim | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qc_check_task | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| qc_check_task | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| qc_check_task | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/terra-tools:2024-08-27 | Optional |
| qc_check_task | gambit_predicted_taxon | String | Internal component, do not modify | Optional | |
| qc_check_task | irma_qc_table | File | Internal component, do not modify | Optional | |
| qc_check_task | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qualimap | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| qualimap | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| qualimap | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/qualimap-custom-html:2.3 | Optional |
| qualimap | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_cpu | Int | Number of CPUs to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | artic_guppyplex_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/artic:1.9.0 | Optional |
| read_QC_trim_ont | artic_guppyplex_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| read_QC_trim_ont | genome_length | Int | Length of the genome | 5000000 | Optional |
| read_QC_trim_ont | max_length | Int | Internal component, do not modify | 700 | Optional |
| read_QC_trim_ont | metabuli_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | metabuli_db | File | Metabuli database for read taxonomy classification, compressed in .tar.gz format | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/refseq_virus-v223.tar.gz | Optional |
| read_QC_trim_ont | metabuli_disk_size | Int | Amount of storage (in GB) to allocate to the task | 250 | Optional |
| read_QC_trim_ont | metabuli_docker_image | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/metabuli:1.1.1 | Optional |
| read_QC_trim_ont | metabuli_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_ont | metabuli_taxdump_path | File | Path to taxonkit-generated taxdump for Metabuli taxonomy parsing | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/ncbi_taxdump_20260211.tar.gz | Optional |
| read_QC_trim_ont | min_length | Int | Internal component, do not modify | 400 | Optional |
| read_QC_trim_ont | nanoq_cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | nanoq_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/nanoq:0.10.0--hc1c3326_4 | Optional |
| read_QC_trim_ont | nanoq_max_read_length | Int | Maximum read length to use for filtering | 100000 | Optional |
| read_QC_trim_ont | nanoq_max_read_qual | Int | Maximum read quality to use for filtering | 100 | Optional |
| read_QC_trim_ont | nanoq_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_min_read_length | Int | Minimum read length to use for filtering | 500 | Optional |
| read_QC_trim_ont | nanoq_min_read_qual | Int | Minimum read quality to use for filtering | 10 | Optional |
| read_QC_trim_ont | ncbi_scrub_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | ncbi_scrub_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | ncbi_scrub_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/ncbi/sra-human-scrubber:2.2.1 | Optional |
| read_QC_trim_ont | ncbi_scrub_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_ont | rasusa_downsampling_coverage | Float | Internal component, do not modify | 150 | Optional |
| read_QC_trim_ont | rasusa_fraction_of_reads | Float | Subsample to a fraction of the reads - e.g., 0.5 samples half the reads | Optional | |
| read_QC_trim_ont | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_num_bases | String | Explicitly set the number of bases required e.g., 4.3kb, 7Tb, 9000, 4.1MB. If this option is given, --coverage and --genome-size are ignored | Optional | |
| read_QC_trim_ont | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_ont | rasusa_seed | Int | Random seed to use | Optional | |
| read_QC_trim_ont | run_prefix | String | Internal component, do not modify | artic_ncov2019 | Optional |
| read_QC_trim_pe | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_pe | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_pe | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_pe | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_pe | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_pe | expected_contaminants | String | Internal component, do not modify | Optional | |
| read_QC_trim_pe | fastp_args | String | Additional arguments to use with fastp | --detect_adapter_for_pe -g -5 20 -3 20 | Optional |
| read_QC_trim_pe | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_pe | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | max_unexpected_contaminants | Int | Maximum unexpected sequences detected from contaminant FASTA to pass status check | 0 | Optional |
| read_QC_trim_pe | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_pe | min_contaminant_coverage | Float | Minimum breadth of coverage to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_depth | Int | Minimum depth to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_reads_mapped | Int | Minimum number of reads mapped to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_expected_contaminants | Int | Minimum expected sequences detected from contaminant FASTA to pass | Optional | |
| read_QC_trim_pe | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_pe | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_pe | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_pe | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_pe | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_pe | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_pe | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_pe | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_pe | read_decontaminate_fasta | File | FASTA of contaminat sequences to map and remove reads against | Optional | |
| read_QC_trim_pe | read_decontaminate_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_pe | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_pe | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_pe | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_pe | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| read_QC_trim_se | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_se | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_se | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_se | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_se | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_se | fastp_args | String | Additional arguments to use with fastp | -g -5 20 -3 20 | Optional |
| read_QC_trim_se | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_se | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_se | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_se | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_se | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_se | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_se | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_se | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_se | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_se | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_se | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_se | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_se | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_se | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_se | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_se | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| sam_to_sorted_bam | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| sam_to_sorted_bam | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| sam_to_sorted_bam | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/samtools:1.17 | Optional |
| sam_to_sorted_bam | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| sam_to_sorted_bam | min_qual | Int | Minimum quality score for reads to be included in the analysis | Optional | |
| version_capture | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/alpine-plus-bash:3.20.0 | Optional |
| version_capture | timezone | String | Set the time zone to get an accurate date of analysis (uses UTC by default) | Optional |
| Terra Task Name | Variable | Type | Description | Default Value | Terra Status |
|---|---|---|---|---|---|
| freyja_fastq | read1 | File | FASTQ file containing read1 sequences (Illumina or (ONT) | Required | |
| freyja_fastq | reference_genome | File | The reference genome to use; should match the reference used for alignment (Wuhan-Hu-1) | Required | |
| freyja_fastq | samplename | String | The name of the sample being analyzed | Required | |
| freyja_fastq | freyja_lineage_metadata | File | File containing the lineage metadata; the "curated_lineages.json" file found https://github.com/andersen-lab/Freyja/tree/main/freyja/data can be used for this variable. Does not need to be provided if update_db is true or if the freyja_pathogen is provided. | Optional, Required | |
| bwa | cpu | Int | Number of CPUs to allocate to the task | 6 | Optional |
| bwa | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| bwa | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| bwa | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| freyja | adapt | Float | adaptive lasso penalty parameter | 0.0 | Optional |
| freyja | auto_adapt | Boolean | When set to true will use error profile to set adapt value | False | Optional |
| freyja | bootstrap | Boolean | Perform bootstrapping | False | Optional |
| freyja | confirmed_only | Boolean | Include only confirmed SARS-CoV-2 lineages | False | Optional |
| freyja | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| freyja | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/freyja:2.0.1 | Optional |
| freyja | eps | Float | The minimum lineage abundance cut-off value | 0.001 | Optional |
| freyja | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| freyja | number_bootstraps | Int | The number of bootstraps to perform (only used if bootstrap = true) | 100 | Optional |
| freyja | update_db | Boolean | Updates the Freyja reference files (the usher barcodes and lineage metadata files) but will not save them as output (use Freyja_Update for that purpose). If set to true, the freyja_lineage_metadata and freyja_barcodes files are not required. | False | Optional |
| freyja_fastq | collection_date | String | Collection date of wastewater sample (YYYY-MM-DD) | Optional | |
| freyja_fastq | collection_site | String | Collection site of wastewater sample | Optional | |
| freyja_fastq | depth_cutoff | Int | The minimum coverage depth with which to exclude sites below this value and group identical barcodes -- THIS MAY NOT WORK FOR NON-SARS-COV-2 ORGANISMS! | Optional | |
| freyja_fastq | freyja_barcodes | File | Custom barcode file. Does not need to be provided if update_db is true if the freyja_pathogen is provided. | Optional | |
| freyja_fastq | freyja_long_format_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/freyja-microreact:1.0.2 | Optional |
| freyja_fastq | freyja_min_coverage | Int | Minimum genome coverage threshold (--mincov) used by freyja_to_long.py when generating the freyja_parsed_format_tsv output | 60 | Optional |
| freyja_fastq | freyja_pathogen | String | Pathogen to be used by Freyja | SARS-CoV-2 | Optional |
| freyja_fastq | kraken2_target_organism | String | The organism whose abundance the user wants to check in their reads. This should be a proper taxonomic name recognized by the Kraken database. | Severe acute respiratory syndrome coronavirus 2 | Optional |
| freyja_fastq | latitude | Float | Latitude of wastewater sample collection site | Optional | |
| freyja_fastq | longitude | Float | Longitude of wastewater sample collection site | Optional | |
| freyja_fastq | ont | Boolean | Indicates if the input data is derived from an ONT instrument. | False | Optional |
| freyja_fastq | primer_bed | File | The bed file containing the primers used when sequencing was performed | Optional | |
| freyja_fastq | qc_check_table | File | TSV containing values to check quality control metrics | Optional | |
| freyja_fastq | read2 | File | Illumina reverse read file in FASTQ file format (compression optional) | Optional | |
| freyja_fastq | reference_gff | File | The GFF file for reference; should match the reference used for alignment (Wuhan-Hu-1) | Optional | |
| freyja_fastq | run_qualimap | Boolean | When set to true, will run qualimap and provide custom visuals | True | Optional |
| freyja_fastq | sc2_gene_bed | File | BED file depicting SARS-CoV-2 region / gene coordinates | gs://theiagen-public-resources-rp/reference_data/viral/sars-cov-2/sc2_gene_locations.bed | Optional |
| freyja_fastq | trimmomatic_min_length | Int | The minimum length cut-off when performing read cleaning | 25 | Optional |
| freyja_long_format | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja_long_format | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| freyja_long_format | group_by | String | Whether to group samples by collection date or week, options are "date" or "week" | Optional | |
| freyja_long_format | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 4 | Optional |
| gene_coverage | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| gene_coverage | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| gene_coverage | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/pysam:1.23.1 | Optional |
| gene_coverage | exact_match | Boolean | Search for exact matches to query_genes / BED file gene queries | False | Optional |
| gene_coverage | feature_qualifier | String | Feature qualifier to use in parsing GBFF for coverage calculations | product | Optional |
| gene_coverage | feature_type | String | Feature type to use in parsing GBFF for coverage calculations | CDS | Optional |
| gene_coverage | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| gene_coverage | min_depth | Int | Minimum depth to call a base covered | 10 | Optional |
| gene_coverage | min_quality | Int | Minimum base quality to call a base covered | 0 | Optional |
| gene_coverage | query_genes | String | Comma-separated list of query genes to parse GBFF/BED file for region coordinates | Optional | |
| gene_coverage | reference_gbff | File | GenBank flat file depicting the reference genome | Optional | |
| get_fasta_genome_size | cpu | Int | Number of CPUs to allocate to the task | 1 | Optional |
| get_fasta_genome_size | disk_size | Int | Amount of storage (in GB) to allocate to the task | 10 | Optional |
| get_fasta_genome_size | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/seqkit:2.4.0--h9ee0642_0 | Optional |
| get_fasta_genome_size | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| minimap2 | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| minimap2 | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| minimap2 | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/minimap2:2.22 | Optional |
| minimap2 | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| minimap2 | query2 | File | Internal component, do not modify | Optional | |
| nanoplot_clean | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_clean | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_clean | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_clean | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_clean | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| nanoplot_raw | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_raw | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_raw | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_raw | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_raw | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| primer_trim | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| primer_trim | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| primer_trim | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| primer_trim | keep_noprimer_reads | Boolean | Include reads with no primers | True | Optional |
| primer_trim | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qc_check_task | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| qc_check_task | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| qc_check_task | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/terra-tools:2024-08-27 | Optional |
| qc_check_task | gambit_predicted_taxon | String | Internal component, do not modify | Optional | |
| qc_check_task | irma_qc_table | File | Internal component, do not modify | Optional | |
| qc_check_task | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qualimap | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| qualimap | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| qualimap | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/qualimap-custom-html:2.3 | Optional |
| qualimap | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_cpu | Int | Number of CPUs to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | artic_guppyplex_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/artic:1.9.0 | Optional |
| read_QC_trim_ont | artic_guppyplex_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| read_QC_trim_ont | genome_length | Int | Length of the genome | 5000000 | Optional |
| read_QC_trim_ont | max_length | Int | Internal component, do not modify | 700 | Optional |
| read_QC_trim_ont | metabuli_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | metabuli_db | File | Metabuli database for read taxonomy classification, compressed in .tar.gz format | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/refseq_virus-v223.tar.gz | Optional |
| read_QC_trim_ont | metabuli_disk_size | Int | Amount of storage (in GB) to allocate to the task | 250 | Optional |
| read_QC_trim_ont | metabuli_docker_image | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/metabuli:1.1.1 | Optional |
| read_QC_trim_ont | metabuli_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_ont | metabuli_taxdump_path | File | Path to taxonkit-generated taxdump for Metabuli taxonomy parsing | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/ncbi_taxdump_20260211.tar.gz | Optional |
| read_QC_trim_ont | min_length | Int | Internal component, do not modify | 400 | Optional |
| read_QC_trim_ont | nanoq_cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | nanoq_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/nanoq:0.10.0--hc1c3326_4 | Optional |
| read_QC_trim_ont | nanoq_max_read_length | Int | Maximum read length to use for filtering | 100000 | Optional |
| read_QC_trim_ont | nanoq_max_read_qual | Int | Maximum read quality to use for filtering | 100 | Optional |
| read_QC_trim_ont | nanoq_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_min_read_length | Int | Minimum read length to use for filtering | 500 | Optional |
| read_QC_trim_ont | nanoq_min_read_qual | Int | Minimum read quality to use for filtering | 10 | Optional |
| read_QC_trim_ont | ncbi_scrub_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | ncbi_scrub_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | ncbi_scrub_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/ncbi/sra-human-scrubber:2.2.1 | Optional |
| read_QC_trim_ont | ncbi_scrub_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_ont | rasusa_downsampling_coverage | Float | Internal component, do not modify | 150 | Optional |
| read_QC_trim_ont | rasusa_fraction_of_reads | Float | Subsample to a fraction of the reads - e.g., 0.5 samples half the reads | Optional | |
| read_QC_trim_ont | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_num_bases | String | Explicitly set the number of bases required e.g., 4.3kb, 7Tb, 9000, 4.1MB. If this option is given, --coverage and --genome-size are ignored | Optional | |
| read_QC_trim_ont | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_ont | rasusa_seed | Int | Random seed to use | Optional | |
| read_QC_trim_ont | run_prefix | String | Internal component, do not modify | artic_ncov2019 | Optional |
| read_QC_trim_pe | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_pe | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_pe | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_pe | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_pe | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_pe | expected_contaminants | String | Internal component, do not modify | Optional | |
| read_QC_trim_pe | fastp_args | String | Additional arguments to use with fastp | --detect_adapter_for_pe -g -5 20 -3 20 | Optional |
| read_QC_trim_pe | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_pe | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | max_unexpected_contaminants | Int | Maximum unexpected sequences detected from contaminant FASTA to pass status check | 0 | Optional |
| read_QC_trim_pe | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_pe | min_contaminant_coverage | Float | Minimum breadth of coverage to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_depth | Int | Minimum depth to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_reads_mapped | Int | Minimum number of reads mapped to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_expected_contaminants | Int | Minimum expected sequences detected from contaminant FASTA to pass | Optional | |
| read_QC_trim_pe | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_pe | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_pe | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_pe | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_pe | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_pe | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_pe | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_pe | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_pe | read_decontaminate_fasta | File | FASTA of contaminat sequences to map and remove reads against | Optional | |
| read_QC_trim_pe | read_decontaminate_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_pe | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_pe | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_pe | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_pe | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| read_QC_trim_se | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_se | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_se | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_se | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_se | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_se | fastp_args | String | Additional arguments to use with fastp | -g -5 20 -3 20 | Optional |
| read_QC_trim_se | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_se | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_se | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_se | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_se | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_se | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_se | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_se | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_se | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_se | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_se | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_se | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_se | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_se | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_se | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_se | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| sam_to_sorted_bam | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| sam_to_sorted_bam | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| sam_to_sorted_bam | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/samtools:1.17 | Optional |
| sam_to_sorted_bam | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| sam_to_sorted_bam | min_qual | Int | Minimum quality score for reads to be included in the analysis | Optional | |
| version_capture | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/alpine-plus-bash:3.20.0 | Optional |
| version_capture | timezone | String | Set the time zone to get an accurate date of analysis (uses UTC by default) | Optional |
| Terra Task Name | Variable | Type | Description | Default Value | Terra Status |
|---|---|---|---|---|---|
| freyja_fastq | read1 | File | FASTQ file containing read1 sequences (Illumina or (ONT) | Required | |
| freyja_fastq | reference_genome | File | The reference genome to use; should match the reference used for alignment (Wuhan-Hu-1) | Required | |
| freyja_fastq | samplename | String | The name of the sample being analyzed | Required | |
| freyja_fastq | freyja_lineage_metadata | File | File containing the lineage metadata; the "curated_lineages.json" file found https://github.com/andersen-lab/Freyja/tree/main/freyja/data can be used for this variable. Does not need to be provided if update_db is true or if the freyja_pathogen is provided. | Optional, Required | |
| bwa | cpu | Int | Number of CPUs to allocate to the task | 6 | Optional |
| bwa | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| bwa | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| bwa | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| freyja | adapt | Float | adaptive lasso penalty parameter | 0.0 | Optional |
| freyja | auto_adapt | Boolean | When set to true will use error profile to set adapt value | False | Optional |
| freyja | bootstrap | Boolean | Perform bootstrapping | False | Optional |
| freyja | confirmed_only | Boolean | Include only confirmed SARS-CoV-2 lineages | False | Optional |
| freyja | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| freyja | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/freyja:2.0.1 | Optional |
| freyja | eps | Float | The minimum lineage abundance cut-off value | 0.001 | Optional |
| freyja | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| freyja | number_bootstraps | Int | The number of bootstraps to perform (only used if bootstrap = true) | 100 | Optional |
| freyja | update_db | Boolean | Updates the Freyja reference files (the usher barcodes and lineage metadata files) but will not save them as output (use Freyja_Update for that purpose). If set to true, the freyja_lineage_metadata and freyja_barcodes files are not required. | False | Optional |
| freyja_fastq | collection_date | String | Collection date of wastewater sample (YYYY-MM-DD) | Optional | |
| freyja_fastq | collection_site | String | Collection site of wastewater sample | Optional | |
| freyja_fastq | depth_cutoff | Int | The minimum coverage depth with which to exclude sites below this value and group identical barcodes -- THIS MAY NOT WORK FOR NON-SARS-COV-2 ORGANISMS! | Optional | |
| freyja_fastq | freyja_barcodes | File | Custom barcode file. Does not need to be provided if update_db is true if the freyja_pathogen is provided. | Optional | |
| freyja_fastq | freyja_long_format_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/freyja-microreact:1.0.2 | Optional |
| freyja_fastq | freyja_min_coverage | Int | Minimum genome coverage threshold (--mincov) used by freyja_to_long.py when generating the freyja_parsed_format_tsv output | 60 | Optional |
| freyja_fastq | freyja_pathogen | String | Pathogen to be used by Freyja | SARS-CoV-2 | Optional |
| freyja_fastq | kraken2_target_organism | String | The organism whose abundance the user wants to check in their reads. This should be a proper taxonomic name recognized by the Kraken database. | Severe acute respiratory syndrome coronavirus 2 | Optional |
| freyja_fastq | latitude | Float | Latitude of wastewater sample collection site | Optional | |
| freyja_fastq | longitude | Float | Longitude of wastewater sample collection site | Optional | |
| freyja_fastq | ont | Boolean | Indicates if the input data is derived from an ONT instrument. | False | Optional |
| freyja_fastq | primer_bed | File | The bed file containing the primers used when sequencing was performed | Optional | |
| freyja_fastq | qc_check_table | File | TSV containing values to check quality control metrics | Optional | |
| freyja_fastq | read2 | File | Illumina reverse read file in FASTQ file format (compression optional) | Optional | |
| freyja_fastq | reference_gff | File | The GFF file for reference; should match the reference used for alignment (Wuhan-Hu-1) | Optional | |
| freyja_fastq | run_qualimap | Boolean | When set to true, will run qualimap and provide custom visuals | True | Optional |
| freyja_fastq | sc2_gene_bed | File | BED file depicting SARS-CoV-2 region / gene coordinates | gs://theiagen-public-resources-rp/reference_data/viral/sars-cov-2/sc2_gene_locations.bed | Optional |
| freyja_fastq | trimmomatic_min_length | Int | The minimum length cut-off when performing read cleaning | 25 | Optional |
| freyja_long_format | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja_long_format | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| freyja_long_format | group_by | String | Whether to group samples by collection date or week, options are "date" or "week" | Optional | |
| freyja_long_format | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 4 | Optional |
| gene_coverage | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| gene_coverage | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| gene_coverage | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/pysam:1.23.1 | Optional |
| gene_coverage | exact_match | Boolean | Search for exact matches to query_genes / BED file gene queries | False | Optional |
| gene_coverage | feature_qualifier | String | Feature qualifier to use in parsing GBFF for coverage calculations | product | Optional |
| gene_coverage | feature_type | String | Feature type to use in parsing GBFF for coverage calculations | CDS | Optional |
| gene_coverage | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| gene_coverage | min_depth | Int | Minimum depth to call a base covered | 10 | Optional |
| gene_coverage | min_quality | Int | Minimum base quality to call a base covered | 0 | Optional |
| gene_coverage | query_genes | String | Comma-separated list of query genes to parse GBFF/BED file for region coordinates | Optional | |
| gene_coverage | reference_gbff | File | GenBank flat file depicting the reference genome | Optional | |
| get_fasta_genome_size | cpu | Int | Number of CPUs to allocate to the task | 1 | Optional |
| get_fasta_genome_size | disk_size | Int | Amount of storage (in GB) to allocate to the task | 10 | Optional |
| get_fasta_genome_size | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/seqkit:2.4.0--h9ee0642_0 | Optional |
| get_fasta_genome_size | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| minimap2 | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| minimap2 | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| minimap2 | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/minimap2:2.22 | Optional |
| minimap2 | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| minimap2 | query2 | File | Internal component, do not modify | Optional | |
| nanoplot_clean | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_clean | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_clean | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_clean | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_clean | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| nanoplot_raw | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| nanoplot_raw | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| nanoplot_raw | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/nanoplot:1.40.0 | Optional |
| nanoplot_raw | max_length | Int | The maximum length of clean reads, for which reads longer than the length specified will be hidden. | 100000 | Optional |
| nanoplot_raw | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| primer_trim | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| primer_trim | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| primer_trim | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/ivar:1.3.1-titan | Optional |
| primer_trim | keep_noprimer_reads | Boolean | Include reads with no primers | True | Optional |
| primer_trim | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qc_check_task | cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| qc_check_task | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| qc_check_task | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/terra-tools:2024-08-27 | Optional |
| qc_check_task | gambit_predicted_taxon | String | Internal component, do not modify | Optional | |
| qc_check_task | irma_qc_table | File | Internal component, do not modify | Optional | |
| qc_check_task | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| qualimap | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| qualimap | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| qualimap | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/qualimap-custom-html:2.3 | Optional |
| qualimap | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_cpu | Int | Number of CPUs to allocate to the task | 8 | Optional |
| read_QC_trim_ont | artic_guppyplex_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | artic_guppyplex_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/artic:1.9.0 | Optional |
| read_QC_trim_ont | artic_guppyplex_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 16 | Optional |
| read_QC_trim_ont | genome_length | Int | Length of the genome | 5000000 | Optional |
| read_QC_trim_ont | max_length | Int | Internal component, do not modify | 700 | Optional |
| read_QC_trim_ont | metabuli_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | metabuli_db | File | Metabuli database for read taxonomy classification, compressed in .tar.gz format | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/refseq_virus-v223.tar.gz | Optional |
| read_QC_trim_ont | metabuli_disk_size | Int | Amount of storage (in GB) to allocate to the task | 250 | Optional |
| read_QC_trim_ont | metabuli_docker_image | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/metabuli:1.1.1 | Optional |
| read_QC_trim_ont | metabuli_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_ont | metabuli_taxdump_path | File | Path to taxonkit-generated taxdump for Metabuli taxonomy parsing | gs://theiagen-public-resources-rp/reference_data/databases/metabuli/ncbi_taxdump_20260211.tar.gz | Optional |
| read_QC_trim_ont | min_length | Int | Internal component, do not modify | 400 | Optional |
| read_QC_trim_ont | nanoq_cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | nanoq_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/biocontainers/nanoq:0.10.0--hc1c3326_4 | Optional |
| read_QC_trim_ont | nanoq_max_read_length | Int | Maximum read length to use for filtering | 100000 | Optional |
| read_QC_trim_ont | nanoq_max_read_qual | Int | Maximum read quality to use for filtering | 100 | Optional |
| read_QC_trim_ont | nanoq_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| read_QC_trim_ont | nanoq_min_read_length | Int | Minimum read length to use for filtering | 500 | Optional |
| read_QC_trim_ont | nanoq_min_read_qual | Int | Minimum read quality to use for filtering | 10 | Optional |
| read_QC_trim_ont | ncbi_scrub_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | ncbi_scrub_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | ncbi_scrub_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/ncbi/sra-human-scrubber:2.2.1 | Optional |
| read_QC_trim_ont | ncbi_scrub_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_ont | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_ont | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_ont | rasusa_downsampling_coverage | Float | Internal component, do not modify | 150 | Optional |
| read_QC_trim_ont | rasusa_fraction_of_reads | Float | Subsample to a fraction of the reads - e.g., 0.5 samples half the reads | Optional | |
| read_QC_trim_ont | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_ont | rasusa_num_bases | String | Explicitly set the number of bases required e.g., 4.3kb, 7Tb, 9000, 4.1MB. If this option is given, --coverage and --genome-size are ignored | Optional | |
| read_QC_trim_ont | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_ont | rasusa_seed | Int | Random seed to use | Optional | |
| read_QC_trim_ont | run_prefix | String | Internal component, do not modify | artic_ncov2019 | Optional |
| read_QC_trim_pe | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_pe | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_pe | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_pe | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_pe | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_pe | expected_contaminants | String | Internal component, do not modify | Optional | |
| read_QC_trim_pe | fastp_args | String | Additional arguments to use with fastp | --detect_adapter_for_pe -g -5 20 -3 20 | Optional |
| read_QC_trim_pe | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_pe | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | max_unexpected_contaminants | Int | Maximum unexpected sequences detected from contaminant FASTA to pass status check | 0 | Optional |
| read_QC_trim_pe | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_pe | min_contaminant_coverage | Float | Minimum breadth of coverage to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_depth | Int | Minimum depth to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_contaminant_reads_mapped | Int | Minimum number of reads mapped to identify a contaminant sequence within the status check (does not affect read cleaning) | 0 | Optional |
| read_QC_trim_pe | min_expected_contaminants | Int | Minimum expected sequences detected from contaminant FASTA to pass | Optional | |
| read_QC_trim_pe | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_pe | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_pe | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_pe | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_pe | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_pe | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_pe | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_pe | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_pe | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_pe | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_pe | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_pe | read_decontaminate_fasta | File | FASTA of contaminat sequences to map and remove reads against | Optional | |
| read_QC_trim_pe | read_decontaminate_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_pe | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_pe | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_pe | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_pe | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_pe | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| read_QC_trim_se | adapters | File | A FASTA file containing adapter sequences | Optional | |
| read_QC_trim_se | bbduk_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | bracken_kmer_length | Int | Kmer length for Bracken to use instead of auto-detection - must be present in database | Optional | |
| read_QC_trim_se | call_bracken | Boolean | Call Bracken kraken2 report refinement | True | Optional |
| read_QC_trim_se | call_midas | Boolean | True/False variable that determines if the MIDAS task should be called. | False | Optional |
| read_QC_trim_se | call_rasusa | Boolean | True/False variable that determines if the RASUSA task should be called. | False | Optional |
| read_QC_trim_se | fastp_args | String | Additional arguments to use with fastp | -g -5 20 -3 20 | Optional |
| read_QC_trim_se | kraken_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | kraken_db | File | A kraken2 database to use with the kraken2 optional task. The file must be a .tar.gz kraken2 database. | gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz | Optional |
| read_QC_trim_se | kraken_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | kraken_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 32 | Optional |
| read_QC_trim_se | midas_db | File | Database to use with MIDAS. Not required as one will be auto-selected when running the MIDAS task. | gs://theiagen-public-resources-rp/reference_data/databases/midas/midas_db_v1.2.tar.gz | Optional |
| read_QC_trim_se | phix | File | The file containing the phix sequence to be used during bbduk task | Optional | |
| read_QC_trim_se | rasusa_cpu | Int | Number of CPUs to allocate to the task | 4 | Optional |
| read_QC_trim_se | rasusa_disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| read_QC_trim_se | rasusa_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/rasusa:2.1.0 | Optional |
| read_QC_trim_se | rasusa_downsampling_coverage | Float | The desired coverage to sub-sample the reads to with RASUSA | 150 | Optional |
| read_QC_trim_se | rasusa_fraction_of_reads | Float | The fraction of reads to retain during downsampling | Optional | |
| read_QC_trim_se | rasusa_genome_length | String | The length of the genome to use for downsampling calculations | Optional | |
| read_QC_trim_se | rasusa_memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| read_QC_trim_se | rasusa_num_bases | String | The bases to use for downsampling with RASUSA | Optional | |
| read_QC_trim_se | rasusa_num_reads | Int | Subsample to a specific number of reads | Optional | |
| read_QC_trim_se | rasusa_seed | Int | Random seed for reproducibility | Optional | |
| read_QC_trim_se | read_processing | String | Options: "trimmomatic" or "fastp" to indicate which read trimming module to use | trimmomatic | Optional |
| read_QC_trim_se | read_qc | String | Allows the user to decide between fastq_scan (default) and fastqc for the evaluation of read quality. | fastq_scan | Optional |
| read_QC_trim_se | trim_quality_min_score | Int | The minimum quality score to keep during trimming | 30 | Optional |
| read_QC_trim_se | trim_window_size | Int | The window size to use during trimming | 4 | Optional |
| read_QC_trim_se | trimmomatic_override_args | String | Additional arguments to pass to trimmomatic. Can be used to override all trimming parameters | Optional | |
| sam_to_sorted_bam | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| sam_to_sorted_bam | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| sam_to_sorted_bam | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/samtools:1.17 | Optional |
| sam_to_sorted_bam | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 8 | Optional |
| sam_to_sorted_bam | min_qual | Int | Minimum quality score for reads to be included in the analysis | Optional | |
| version_capture | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/alpine-plus-bash:3.20.0 | Optional |
| version_capture | timezone | String | Set the time zone to get an accurate date of analysis (uses UTC by default) | Optional |
Analysis Tasks¶
read_QC_trim: Read Quality Trimming, Adapter Removal, Quantification, and Identification
read_QC_trim is a sub-workflow that removes low-quality reads, low-quality regions of reads, and sequencing adapters to improve data quality. It uses a number of tasks, described below. The differences between the PE and SE versions of the read_QC_trim sub-workflow lie in the default parameters, the use of two or one input read file(s), and the different output files.
By default, read_qc is set to "fastq_scan". To use FastQC instead, set read_qc to "fastqc". These tasks are mutually exclusive.
fastq-scan: Read Quantification (default)
Read quantification is available via fastq-scan by default.
fastq-scan quantifies the forward and reverse reads in FASTQ files. For paired-end data, it also provides the total number of read pairs. This task is run once with raw reads as input and once with clean reads as input. If QC has been performed correctly, you should expect fewer clean reads than raw reads.
fastq-scan Technical Details
| Links | |
|---|---|
| Task | task_fastq_scan.wdl |
| Software Source Code | fastq-scan on GitHub |
| Software Documentation | fastq-scan on GitHub |
FastQC: Read Quantification (alternative)
To activate this task, set read_qc to "fastqc".
FastQC quantifies the forward and reverse reads in FASTQ files. For paired-end data, it also provides the total number of read pairs. This task is run once with raw reads as input and once with clean reads as input. If QC has been performed correctly, you should expect fewer clean reads than raw reads.
This tool also provides a graphical visualization of the read quality.
FastQC Technical Details
| Links | |
|---|---|
| Task | task_fastqc.wdl |
| Software Source Code | FastQC on Github |
| Software Documentation | FastQC Website |
read_decontaminate: Mapping-based Read Decontamination (optional)
Activate this task by providing a read_decontaminate_fasta.
Known contaminant genetic data can be removed by mapping directly to an input read_decontaminate_fasta. This input can be a host genome, common microbial contaminant genome, or intentionally spiked sequences. The mapping statistics and aligned reads to the contaminant FASTA are output in JSON-formatted mappings, while downstream quality control tasks will input the decontaminated reads. An optional "pass/fail" status can be output based on identification of expected/unexpected sequences if the expected_contaminants input is populated with a comma-delimited string of expected sequence headers - expected_contaminants must exactly match sequence headers in the input.
The detailed steps and tasks are as follows:
Minimap2: Read Alignment
Minimap2 is a popular aligner that is used to align reads (or assemblies) to an assembly file. In Minimap2, "modes" are a group of preset options.
The mode used in this task is map-ont which is the default mode for long reads and indicates that long reads of ~10% error rates should be aligned to the reference genome. The output file is in SAM format.
For more information regarding modes and the available options for Minimap2, please see the Minimap2 manpage.
Minimap2 Technical Details
| Links | |
|---|---|
| Task | task_minimap2.wdl |
| Software Source Code | Minimap2 on GitHub |
| Software Documentation | Minimap2 |
| Original Publication(s) | Minimap2: pairwise alignment for nucleotide sequences |
parse_mapping: Extract Unaligned Reads
The bam_to_unaligned_fastq sub-task will extract a FASTQ file of reads that failed to align, while removing unpaired reads.
Parse Mapping Technical Details
| Links | |
|---|---|
| Task | task_parse_mapping.wdl |
| Software Source Code | samtools on GitHub |
| Software Documentation | samtools |
| Original Publication(s) | The Sequence Alignment/Map format and SAMtools Twelve Years of SAMtools and BCFtools |
mapping_stats: Read Mapping Statistics
The Read Mapping Statistics task generates mapping statistics from a BAM file. It uses samtools to generate a summary of the mapping statistics, which includes coverage, depth, average base quality, average mapping quality, and other relevant metrics. These statistics are also reported on a per sequence basis.
Read Mapping Statistics Technical Details
| Links | |
|---|---|
| Task | task_mapping_stats.wdl |
| Software Source Code | samtools on GitHub |
| Software Documentation | samtools |
| Original Publication(s) | The Sequence Alignment/Map format and SAMtools Twelve Years of SAMtools and BCFtools |
Contaminant_Check: Contaminant Sequence Status Check
The Contaminant Check task outputs a pass/fail status based on if contaminant/host sequences pass thresholds for breadth of coverage, depth of coverage, and number of reads mapped. This task is activated by inputting a comma-delimited string of expected_sequences, which match sequence headers in the provided contaminant/host FASTA. Each sequence from the previously provided contaminant/host FASTA is checked for sufficient read mapping statistics.
The composite status, contaminant_check_status, will report "PASS" if expected and unexpected sequences are identified within the min_expected_seq and max_unexpected_seq thresholds; if not, "FAIL ..." is reported depicting which expected_sequences failed and why, along with which unexpected sequences were identified.
Additionally, the coverage, depth, and number of reads mapped are reported in JSON mappings for the sets of expected and unexpected sequences.
Contaminant Check Technical Details
| Links | |
|---|---|
| Task | task_contaminant_check.wdl |
Read Decontaminate Technical Details
| Links | |
|---|---|
| Subworkflow | wf_read_decontaminate.wdl |
HRRT: Human Host Sequence Removal
All reads of human origin are removed, including their mates, by using NCBI's human read removal tool (HRRT).
HRRT is based on the SRA Taxonomy Analysis Tool and employs a k-mer database constructed of k-mers from Eukaryota derived from all human RefSeq records with any k-mers found in non-Eukaryota RefSeq records subtracted from the database.
HRRT Technical Details
| Links | |
|---|---|
| Task | task_ncbi_scrub.wdl |
| Software Source Code | HRRT on GitHub |
| Software Documentation | HRRT on NCBI |
Rasusa: Read Subsampling (optional)
Rasusa is a tool to randomly subsample sequencing reads to a specified coverage without assuming that all reads are of equal length, making it especially suitable for long-read data while still being applicable to short-read data.
The Rasusa task supports four mutually exclusive subsampling modes:
| Mode | Behavior |
|---|---|
--bases |
Subsample to a target number of bases (e.g. 100M, 4.3kb). Overrides coverage. |
--frac |
Subsample to a fraction of the input reads (e.g. 0.5 keeps half). Overrides coverage. |
--num |
Subsample to an explicit number of reads. Overrides coverage. |
--coverage + --genome-size |
Default mode. Subsamples to a target depth using an estimated genome length. |
If more than one of --bases, --frac, or --num is supplied the task will fail with a descriptive error. See inputs section for details on Terra variable names.
To enable/disable this task, set the call_rasusa parameter to true/false. Each workflow has its own default values for Rasusa which can be overridden by the user:
| Workflow | call_rasusa default |
rasusa_downsampling_coverage default |
|---|---|---|
theiaeuk_illumina_pe |
true |
150 |
theiacov_illumina_pe |
false |
2000 |
theiacov_illumina_se |
false |
2000 |
theiaprok_illumina_pe |
false |
150 |
theiaprok_illumina_se |
false |
150 |
Rasusa executes after host-read removal (HRRT, if applicable) and before read trimming (Trimmomatic or fastp). Classification tasks (Kraken2, MIDAS) always run on the original/raw reads regardless of whether call_rasusa is enabled. The downsampled (but un-trimmed) reads are output to the Terra data table as read1_subsampled_raw and read2_subsampled_raw.
When running in coverage mode, it's strongly recommended to manually set the rasusa_genome_length input parameter in order to ensure accurate downsampling. If not provided, rasusa_genome_length falls back to raw_check_reads.est_genome_length from the read_screen task. Oftentimes, this value can overestimate the true genome length, particularly for large, high-coverage FASTQ files. If skip_screen is set to true, you must supply genome_length explicitly or use one of the override modes above.
Non-deterministic output(s)
This task may yield non-deterministic outputs since it performs random subsampling. To ensure reproducibility, set a value for the rasusa_seed optional input variable.
Rasusa Technical Details
| Links | |
|---|---|
| Task | task_rasusa.wdl |
| Software Source Code | Rasusa on GitHub |
| Software Documentation | Rasusa on GitHub |
| Original Publication(s) | Rasusa: Randomly subsample sequencing reads to a specified coverage |
By default, read_processing is set to "trimmomatic". To use fastp instead, set read_processing to "fastp". These tasks are mutually exclusive.
Trimmomatic: Read Trimming (default)
Read processing is available via Trimmomatic by default.
Trimmomatic trims low-quality regions of Illumina paired-end or single-end reads with a sliding window (with a default window size of 4, specified with trim_window_size), cutting once the average quality within the window falls below the trimmomatic_window_quality (default of 30 for both paired-end and single-end). The read is discarded if it is trimmed below trimmomatic_min_length (default of 75 for paired-end, 25 for single-end).
Adapter trimming with Trimmomatic is disabled by default. It can be enabled by setting trimmomatic_trim_adapters=true. When enabled, Trimmomatic uses its default adapter sequences, TruSeq3-PE-2.fa (for paired-end) or TruSeq3-SE.fa (for single-end) with default adapter clipping settings equivalent to:
ILLUMINACLIP:<adapter_fasta>:2:30:10
2 = seed mismatches
30 = palindrome clip threshold
10 = simple clip threshold
Users can optionally provide a custom adapter file or modify adapter trimming parameters using the trimmomatic_adapter_fasta and trimmomatic_adapter_trim_args respectively. See the Trimmomatic adapter documentation for more details. The trimmomatic_adapter_fasta parameter should just include the path to your fasta file. The trimmomatic_adapter_trim_args parameter should only contain the colon-delimited values that come after the adapter fasta file in the ILLUMINACLIP argument. Example usage:
For more advanced configurations, there are options to override the default trimming parameters via trimmomatic_override_args. Note that when using trimmomatic_override_args, the user is responsible for specifying all desired trimming steps and their order, as the default trimming steps will be ignored. See the Trimmomatic documentation for more details on available trimming steps and their parameters.
Advanced Configuration Example Usage:
Trimmomatic Technical Details
| Links | |
|---|---|
| Task | task_trimmomatic.wdl |
| Software Source Code | Trimmomatic on GitHub |
| Software Documentation | Trimmomatic Website |
| Original Publication(s) | Trimmomatic: a flexible trimmer for Illumina sequence data |
fastp: Read Trimming (alternative)
To activate this task, set read_processing to "fastp".
fastp trims low-quality regions of Illumina paired-end or single-end reads with a sliding window (with a default window size of 4 (10 for TheiaEuk), specified with trim_window_size), cutting once the average quality within the window falls below the trim_quality_trim_score (default of 20 for paired-end, 30 for single-end). The read is discarded if it is trimmed below trim_minlen (default of 75 bases for paired-end, 25 for single-end).
fastp also has additional default parameters and features that are not a part of Trimmomatic's default configuration. Please note --disable_adapter_trimming is explicitly needed in fastp_args to disable adapter trimming via fastp.
Default read-trimming parameters
| Parameter | Explanation |
|---|---|
| -g | enables polyG tail trimming |
| -5 20 | enables read end-trimming |
| -3 20 | enables read end-trimming |
| --detect_adapter_for_pe | More sensitively detects adapters for trimming only for paired-end reads |
Additional arguments can be passed using the fastp_args optional parameter. Please reference the fastp GitHub for a comprehensive list of arguments.
fastp Technical Details
| Links | |
|---|---|
| Task | task_fastp.wdl |
| Software Source Code | fastp on GitHub |
| Software Documentation | fastp on GitHub |
| Original Publication(s) | fastp: an ultra-fast all-in-one FASTQ preprocessor |
BBDuk: Adapter Trimming and PhiX Removal
Adapters are manufactured oligonucleotide sequences attached to DNA fragments during the library preparation process. In Illumina sequencing, these adapter sequences are required for attaching reads to flow cells. You can read more about Illumina's adapter-sequence reference. For genome analysis, it's important to remove these sequences since they're not actually from your sample. If you don't remove them, the downstream analysis may be affected.
By default, the BBDuk task will:
-
Repair disordered read pairs (if they exist) so that the first read in
read1is the same mate of the first read inread2. See theRepair Guidefrom the BBTools package. -
Remove PhiX contamination by filtering out all reads that have a 31-mer match to PhiX. PhiX is a viral genome that is often used as a control in Illumina sequencing runs. Removing PhiX sequences helps to ensure that the data reflects only the target organism's genome. By default this task uses the built-in PhiX reference fasta provided with BBTools (see the PhiX reference sequence), but a custom PhiX reference can be provided via the
phix_fastainput parameter. -
Trim adapters with BBDuk ("Bestus Bioinformaticus" Decontamination Using Kmers). By default this uses the built-in adapter sequences provided with BBTools (see the BBTools adapter reference (
adapters.fa)). If you have custom adapter sequences specific to your library preparation, you can provide them via theadapters_fastainput parameter.
BBDuk Technical Details
| Links | |
|---|---|
| Task | task_bbduk.wdl |
| Software Source Code | BBMap on SourceForge |
| Software Documentation | BBDuk Guide (archived) |
Kraken2 + Bracken: Read Classification
Kraken2 is a bioinformatics tool originally designed for metagenomic applications that is database dependent. It has additionally proven valuable for validating taxonomic assignments and checking contamination of single-species (e.g. bacterial isolate, eukaryotic isolate, viral isolate, etc.) whole genome sequence data.
Bracken is a refinement module that improves the resolution of Kraken2 reports.
Kraken2 is run on both the raw and clean reads.
Database-dependent
This workflow automatically uses a viral-specific Kraken2 database. This database was generated in-house from RefSeq's viral sequence collection and human genome GRCh38. It's available at gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz.
Bracken report refinement
Bracken refines the Kraken2 taxon classification report when call_bracken is set to "true" (default). Bracken uses a Bayesian model to probabilistically estimate read abundances at the species/genus-level. Bracken will output a bracken_report that:
- increases report-level classification resolution up to the species level
- decreases resolution of sub-species report-level classifications, e.g. Severe acute respiratory syndrome coronavirus 2 will be grouped into Betacoronavirus pandemicum
- does not affect read-level classification and extraction
- will not be used in downstream
percent_humanandpercent_target_organismcalculations - is provided in place of Kraken reports in downstream tasks, such as
qc_checkandkrona - output separate of the
kraken/kraken2_report
By default, Bracken will reference the k-mer database that is closest to the mean read length of the input. This reference k-mer database size can be directly set using the bracken_kmer_length input, though it MUST correspond to an available k-mer database within the Kraken2 database (named database<KMER_LENGTH>mers.kmer_distrib). Bracken will be skipped if there are no k-mer libraries in the Kraken2 database.
Kraken2 Technical Details
| Links | |
|---|---|
| Task | task_kraken2.wdl |
| Software Source Code | Kraken2 on GitHub Bracken on GitHub |
| Software Documentation | Kraken2 Documentation Bracken Documentation |
| Original Publication(s) | Improved metagenomic analysis with Kraken 2 Bracken: estimating species abundance in metagenomics data |
read_QC_trim Technical Details
| Links | |
|---|---|
| Subworkflow | wf_read_QC_trim_pe.wdl wf_read_QC_trim_se.wdl |
BWA: Read Alignment to the Assembly
BWA (Burrows-Wheeler Aligner) is used to align the cleaned read files to a reference genome provided by the user.
BWA Technical Details
| Links | |
|---|---|
| Task | task_bwa.wdl |
| Software Source Code | BWA on GitHub |
| Software Documentation | BWA Documentation |
| Original Publication(s) | Fast and accurate short read alignment with Burrows-Wheeler transform |
iVar trim: Primer Trimming
The optional input, keep_noprimer_reads, does not have to be modified.
Using the user-provided (or a organism-specific parameters-determined) primer_bed file, iVar soft-clips primer sequences from an aligned and sorted BAM file.
iVar will trim any reads that start or end within the (0-based index) coordinates provided in the BED file. It does not take the sequence of bases itself into account. This allows iVar to accurately trim primer sequences despite potential mismatches between sequencing reads and primer sequences in the aligned region.
Following the trimming of primer sequences, iVar then trims the reads based on a quality threshold of 20 using a sliding window approach (default: 4). If the average base quality drops below the threshold, the remainder of the read is soft-clipped. Reads exceeding the minimum length (default: 30) after trimming are retained in the output BAM file.
iVar Technical Details
| Links | |
|---|---|
| Task | task_ivar_primer_trim.wdl |
| Software Source Code | iVar on GitHub |
| Software Documentation | iVar on GitHub |
| Original Publication(s) | An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar |
QualiMap: BAM File Quality Assessment
QualiMap evaluates the quality of alignment data in BAM files by computing various metrics including coverage distribution, mapping quality, GC content, and various metrics analyzed across the reference. It provides comprehensive quality control reports for next-generation sequencing alignment data.
This task generates both standard QualiMap reports and custom interactive HTML visualizations for genome coverage and mapping quality across the reference sequence. The results are bundled into a compressed archive for easy download and review.
QualiMap Technical Details
| Links | |
|---|---|
| Task | task_qualimap.wdl |
| Software Source Code | QualiMap on Bitbucket |
| Software Documentation | QualiMap Documentation |
| Original Publication(s) | QualiMap: evaluating next-generation sequencing alignment data |
qc_check: Check QC Metrics Against User-Defined Thresholds (optional)
To activate this task, provide a qc_check_table as input.
The QC Check task compares generated QC metrics against user-defined thresholds for each metric. This task will run if the user provides a qc_check_table TSV file. If all QC metrics meet the threshold, the qc_check output variable will read QC_PASS. Otherwise, the output will read QC_NA if the task could not proceed or QC_ALERT followed by a string indicating what metric failed.
Thresholds for percent human classified reads and percent human classified dehosted reads are noted as "classified_human" and "classified_human_dehosted" respectively. In other workflows, these QC check columns are named "kraken_human" or "metabuli_human", but Freyja uses the "classified_" prefix because the classification software used will vary depending on read type (kraken2 for Illumina, Metabuli for ONT).
Formatting the qc_check_table.tsv
- The first column of the qc_check_table lists the
organismthat the task will assess and the header of this column must be "taxon". - Each subsequent column indicates a QC metric and lists a threshold for each organism that will be checked. The column names must exactly match expected values, so we highly recommend copy and pasting the header from the template file below as a starting place.
Template qc_check_table.tsv files
- Freyja_FASTQ: freyja_qc_check_template.tsv
Example Purposes Only
The QC threshold values shown in the file above are for example purposes only and should not be presumed to be sufficient for every dataset.
qc_check Technical Details
| Links | |
|---|---|
| Task | task_qc_check_phb.wdl |
read_QC_trim: Read Quality Trimming, Adapter Removal, Quantification, and Identification
read_QC_trim is a sub-workflow that removes low-quality reads, low-quality regions of reads, and sequencing adapters to improve data quality. It uses a number of tasks, described below. The differences between the PE and SE versions of the read_QC_trim sub-workflow lie in the default parameters, the use of two or one input read file(s), and the different output files.
By default, read_qc is set to "fastq_scan". To use FastQC instead, set read_qc to "fastqc". These tasks are mutually exclusive.
fastq-scan: Read Quantification (default)
Read quantification is available via fastq-scan by default.
fastq-scan quantifies the forward and reverse reads in FASTQ files. For paired-end data, it also provides the total number of read pairs. This task is run once with raw reads as input and once with clean reads as input. If QC has been performed correctly, you should expect fewer clean reads than raw reads.
fastq-scan Technical Details
| Links | |
|---|---|
| Task | task_fastq_scan.wdl |
| Software Source Code | fastq-scan on GitHub |
| Software Documentation | fastq-scan on GitHub |
FastQC: Read Quantification (alternative)
To activate this task, set read_qc to "fastqc".
FastQC quantifies the forward and reverse reads in FASTQ files. For paired-end data, it also provides the total number of read pairs. This task is run once with raw reads as input and once with clean reads as input. If QC has been performed correctly, you should expect fewer clean reads than raw reads.
This tool also provides a graphical visualization of the read quality.
FastQC Technical Details
| Links | |
|---|---|
| Task | task_fastqc.wdl |
| Software Source Code | FastQC on Github |
| Software Documentation | FastQC Website |
read_decontaminate: Mapping-based Read Decontamination (optional)
Activate this task by providing a read_decontaminate_fasta.
Known contaminant genetic data can be removed by mapping directly to an input read_decontaminate_fasta. This input can be a host genome, common microbial contaminant genome, or intentionally spiked sequences. The mapping statistics and aligned reads to the contaminant FASTA are output in JSON-formatted mappings, while downstream quality control tasks will input the decontaminated reads. An optional "pass/fail" status can be output based on identification of expected/unexpected sequences if the expected_contaminants input is populated with a comma-delimited string of expected sequence headers - expected_contaminants must exactly match sequence headers in the input.
The detailed steps and tasks are as follows:
Minimap2: Read Alignment
Minimap2 is a popular aligner that is used to align reads (or assemblies) to an assembly file. In Minimap2, "modes" are a group of preset options.
The mode used in this task is map-ont which is the default mode for long reads and indicates that long reads of ~10% error rates should be aligned to the reference genome. The output file is in SAM format.
For more information regarding modes and the available options for Minimap2, please see the Minimap2 manpage.
Minimap2 Technical Details
| Links | |
|---|---|
| Task | task_minimap2.wdl |
| Software Source Code | Minimap2 on GitHub |
| Software Documentation | Minimap2 |
| Original Publication(s) | Minimap2: pairwise alignment for nucleotide sequences |
parse_mapping: Extract Unaligned Reads
The bam_to_unaligned_fastq sub-task will extract a FASTQ file of reads that failed to align, while removing unpaired reads.
Parse Mapping Technical Details
| Links | |
|---|---|
| Task | task_parse_mapping.wdl |
| Software Source Code | samtools on GitHub |
| Software Documentation | samtools |
| Original Publication(s) | The Sequence Alignment/Map format and SAMtools Twelve Years of SAMtools and BCFtools |
mapping_stats: Read Mapping Statistics
The Read Mapping Statistics task generates mapping statistics from a BAM file. It uses samtools to generate a summary of the mapping statistics, which includes coverage, depth, average base quality, average mapping quality, and other relevant metrics. These statistics are also reported on a per sequence basis.
Read Mapping Statistics Technical Details
| Links | |
|---|---|
| Task | task_mapping_stats.wdl |
| Software Source Code | samtools on GitHub |
| Software Documentation | samtools |
| Original Publication(s) | The Sequence Alignment/Map format and SAMtools Twelve Years of SAMtools and BCFtools |
Contaminant_Check: Contaminant Sequence Status Check
The Contaminant Check task outputs a pass/fail status based on if contaminant/host sequences pass thresholds for breadth of coverage, depth of coverage, and number of reads mapped. This task is activated by inputting a comma-delimited string of expected_sequences, which match sequence headers in the provided contaminant/host FASTA. Each sequence from the previously provided contaminant/host FASTA is checked for sufficient read mapping statistics.
The composite status, contaminant_check_status, will report "PASS" if expected and unexpected sequences are identified within the min_expected_seq and max_unexpected_seq thresholds; if not, "FAIL ..." is reported depicting which expected_sequences failed and why, along with which unexpected sequences were identified.
Additionally, the coverage, depth, and number of reads mapped are reported in JSON mappings for the sets of expected and unexpected sequences.
Contaminant Check Technical Details
| Links | |
|---|---|
| Task | task_contaminant_check.wdl |
Read Decontaminate Technical Details
| Links | |
|---|---|
| Subworkflow | wf_read_decontaminate.wdl |
HRRT: Human Host Sequence Removal
All reads of human origin are removed, including their mates, by using NCBI's human read removal tool (HRRT).
HRRT is based on the SRA Taxonomy Analysis Tool and employs a k-mer database constructed of k-mers from Eukaryota derived from all human RefSeq records with any k-mers found in non-Eukaryota RefSeq records subtracted from the database.
HRRT Technical Details
| Links | |
|---|---|
| Task | task_ncbi_scrub.wdl |
| Software Source Code | HRRT on GitHub |
| Software Documentation | HRRT on NCBI |
Rasusa: Read Subsampling (optional)
Rasusa is a tool to randomly subsample sequencing reads to a specified coverage without assuming that all reads are of equal length, making it especially suitable for long-read data while still being applicable to short-read data.
The Rasusa task supports four mutually exclusive subsampling modes:
| Mode | Behavior |
|---|---|
--bases |
Subsample to a target number of bases (e.g. 100M, 4.3kb). Overrides coverage. |
--frac |
Subsample to a fraction of the input reads (e.g. 0.5 keeps half). Overrides coverage. |
--num |
Subsample to an explicit number of reads. Overrides coverage. |
--coverage + --genome-size |
Default mode. Subsamples to a target depth using an estimated genome length. |
If more than one of --bases, --frac, or --num is supplied the task will fail with a descriptive error. See inputs section for details on Terra variable names.
To enable/disable this task, set the call_rasusa parameter to true/false. Each workflow has its own default values for Rasusa which can be overridden by the user:
| Workflow | call_rasusa default |
rasusa_downsampling_coverage default |
|---|---|---|
theiaeuk_illumina_pe |
true |
150 |
theiacov_illumina_pe |
false |
2000 |
theiacov_illumina_se |
false |
2000 |
theiaprok_illumina_pe |
false |
150 |
theiaprok_illumina_se |
false |
150 |
Rasusa executes after host-read removal (HRRT, if applicable) and before read trimming (Trimmomatic or fastp). Classification tasks (Kraken2, MIDAS) always run on the original/raw reads regardless of whether call_rasusa is enabled. The downsampled (but un-trimmed) reads are output to the Terra data table as read1_subsampled_raw and read2_subsampled_raw.
When running in coverage mode, it's strongly recommended to manually set the rasusa_genome_length input parameter in order to ensure accurate downsampling. If not provided, rasusa_genome_length falls back to raw_check_reads.est_genome_length from the read_screen task. Oftentimes, this value can overestimate the true genome length, particularly for large, high-coverage FASTQ files. If skip_screen is set to true, you must supply genome_length explicitly or use one of the override modes above.
Non-deterministic output(s)
This task may yield non-deterministic outputs since it performs random subsampling. To ensure reproducibility, set a value for the rasusa_seed optional input variable.
Rasusa Technical Details
| Links | |
|---|---|
| Task | task_rasusa.wdl |
| Software Source Code | Rasusa on GitHub |
| Software Documentation | Rasusa on GitHub |
| Original Publication(s) | Rasusa: Randomly subsample sequencing reads to a specified coverage |
By default, read_processing is set to "trimmomatic". To use fastp instead, set read_processing to "fastp". These tasks are mutually exclusive.
Trimmomatic: Read Trimming (default)
Read processing is available via Trimmomatic by default.
Trimmomatic trims low-quality regions of Illumina paired-end or single-end reads with a sliding window (with a default window size of 4, specified with trim_window_size), cutting once the average quality within the window falls below the trimmomatic_window_quality (default of 30 for both paired-end and single-end). The read is discarded if it is trimmed below trimmomatic_min_length (default of 75 for paired-end, 25 for single-end).
Adapter trimming with Trimmomatic is disabled by default. It can be enabled by setting trimmomatic_trim_adapters=true. When enabled, Trimmomatic uses its default adapter sequences, TruSeq3-PE-2.fa (for paired-end) or TruSeq3-SE.fa (for single-end) with default adapter clipping settings equivalent to:
ILLUMINACLIP:<adapter_fasta>:2:30:10
2 = seed mismatches
30 = palindrome clip threshold
10 = simple clip threshold
Users can optionally provide a custom adapter file or modify adapter trimming parameters using the trimmomatic_adapter_fasta and trimmomatic_adapter_trim_args respectively. See the Trimmomatic adapter documentation for more details. The trimmomatic_adapter_fasta parameter should just include the path to your fasta file. The trimmomatic_adapter_trim_args parameter should only contain the colon-delimited values that come after the adapter fasta file in the ILLUMINACLIP argument. Example usage:
For more advanced configurations, there are options to override the default trimming parameters via trimmomatic_override_args. Note that when using trimmomatic_override_args, the user is responsible for specifying all desired trimming steps and their order, as the default trimming steps will be ignored. See the Trimmomatic documentation for more details on available trimming steps and their parameters.
Advanced Configuration Example Usage:
Trimmomatic Technical Details
| Links | |
|---|---|
| Task | task_trimmomatic.wdl |
| Software Source Code | Trimmomatic on GitHub |
| Software Documentation | Trimmomatic Website |
| Original Publication(s) | Trimmomatic: a flexible trimmer for Illumina sequence data |
fastp: Read Trimming (alternative)
To activate this task, set read_processing to "fastp".
fastp trims low-quality regions of Illumina paired-end or single-end reads with a sliding window (with a default window size of 4 (10 for TheiaEuk), specified with trim_window_size), cutting once the average quality within the window falls below the trim_quality_trim_score (default of 20 for paired-end, 30 for single-end). The read is discarded if it is trimmed below trim_minlen (default of 75 bases for paired-end, 25 for single-end).
fastp also has additional default parameters and features that are not a part of Trimmomatic's default configuration. Please note --disable_adapter_trimming is explicitly needed in fastp_args to disable adapter trimming via fastp.
Default read-trimming parameters
| Parameter | Explanation |
|---|---|
| -g | enables polyG tail trimming |
| -5 20 | enables read end-trimming |
| -3 20 | enables read end-trimming |
| --detect_adapter_for_pe | More sensitively detects adapters for trimming only for paired-end reads |
Additional arguments can be passed using the fastp_args optional parameter. Please reference the fastp GitHub for a comprehensive list of arguments.
fastp Technical Details
| Links | |
|---|---|
| Task | task_fastp.wdl |
| Software Source Code | fastp on GitHub |
| Software Documentation | fastp on GitHub |
| Original Publication(s) | fastp: an ultra-fast all-in-one FASTQ preprocessor |
BBDuk: Adapter Trimming and PhiX Removal
Adapters are manufactured oligonucleotide sequences attached to DNA fragments during the library preparation process. In Illumina sequencing, these adapter sequences are required for attaching reads to flow cells. You can read more about Illumina's adapter-sequence reference. For genome analysis, it's important to remove these sequences since they're not actually from your sample. If you don't remove them, the downstream analysis may be affected.
By default, the BBDuk task will:
-
Repair disordered read pairs (if they exist) so that the first read in
read1is the same mate of the first read inread2. See theRepair Guidefrom the BBTools package. -
Remove PhiX contamination by filtering out all reads that have a 31-mer match to PhiX. PhiX is a viral genome that is often used as a control in Illumina sequencing runs. Removing PhiX sequences helps to ensure that the data reflects only the target organism's genome. By default this task uses the built-in PhiX reference fasta provided with BBTools (see the PhiX reference sequence), but a custom PhiX reference can be provided via the
phix_fastainput parameter. -
Trim adapters with BBDuk ("Bestus Bioinformaticus" Decontamination Using Kmers). By default this uses the built-in adapter sequences provided with BBTools (see the BBTools adapter reference (
adapters.fa)). If you have custom adapter sequences specific to your library preparation, you can provide them via theadapters_fastainput parameter.
BBDuk Technical Details
| Links | |
|---|---|
| Task | task_bbduk.wdl |
| Software Source Code | BBMap on SourceForge |
| Software Documentation | BBDuk Guide (archived) |
Kraken2 + Bracken: Read Classification
Kraken2 is a bioinformatics tool originally designed for metagenomic applications that is database dependent. It has additionally proven valuable for validating taxonomic assignments and checking contamination of single-species (e.g. bacterial isolate, eukaryotic isolate, viral isolate, etc.) whole genome sequence data.
Bracken is a refinement module that improves the resolution of Kraken2 reports.
Kraken2 is run on both the raw and clean reads.
Database-dependent
This workflow automatically uses a viral-specific Kraken2 database. This database was generated in-house from RefSeq's viral sequence collection and human genome GRCh38. It's available at gs://theiagen-public-resources-rp/reference_data/databases/kraken2/k2_viral-refseq_human-GRCh38_20260220.tar.gz.
Bracken report refinement
Bracken refines the Kraken2 taxon classification report when call_bracken is set to "true" (default). Bracken uses a Bayesian model to probabilistically estimate read abundances at the species/genus-level. Bracken will output a bracken_report that:
- increases report-level classification resolution up to the species level
- decreases resolution of sub-species report-level classifications, e.g. Severe acute respiratory syndrome coronavirus 2 will be grouped into Betacoronavirus pandemicum
- does not affect read-level classification and extraction
- will not be used in downstream
percent_humanandpercent_target_organismcalculations - is provided in place of Kraken reports in downstream tasks, such as
qc_checkandkrona - output separate of the
kraken/kraken2_report
By default, Bracken will reference the k-mer database that is closest to the mean read length of the input. This reference k-mer database size can be directly set using the bracken_kmer_length input, though it MUST correspond to an available k-mer database within the Kraken2 database (named database<KMER_LENGTH>mers.kmer_distrib). Bracken will be skipped if there are no k-mer libraries in the Kraken2 database.
Kraken2 Technical Details
| Links | |
|---|---|
| Task | task_kraken2.wdl |
| Software Source Code | Kraken2 on GitHub Bracken on GitHub |
| Software Documentation | Kraken2 Documentation Bracken Documentation |
| Original Publication(s) | Improved metagenomic analysis with Kraken 2 Bracken: estimating species abundance in metagenomics data |
read_QC_trim Technical Details
| Links | |
|---|---|
| Subworkflow | wf_read_QC_trim_pe.wdl wf_read_QC_trim_se.wdl |
BWA: Read Alignment to the Assembly
BWA (Burrows-Wheeler Aligner) is used to align the cleaned read files to a reference genome provided by the user.
BWA Technical Details
| Links | |
|---|---|
| Task | task_bwa.wdl |
| Software Source Code | BWA on GitHub |
| Software Documentation | BWA Documentation |
| Original Publication(s) | Fast and accurate short read alignment with Burrows-Wheeler transform |
iVar trim: Primer Trimming
The optional input, keep_noprimer_reads, does not have to be modified.
Using the user-provided (or a organism-specific parameters-determined) primer_bed file, iVar soft-clips primer sequences from an aligned and sorted BAM file.
iVar will trim any reads that start or end within the (0-based index) coordinates provided in the BED file. It does not take the sequence of bases itself into account. This allows iVar to accurately trim primer sequences despite potential mismatches between sequencing reads and primer sequences in the aligned region.
Following the trimming of primer sequences, iVar then trims the reads based on a quality threshold of 20 using a sliding window approach (default: 4). If the average base quality drops below the threshold, the remainder of the read is soft-clipped. Reads exceeding the minimum length (default: 30) after trimming are retained in the output BAM file.
iVar Technical Details
| Links | |
|---|---|
| Task | task_ivar_primer_trim.wdl |
| Software Source Code | iVar on GitHub |
| Software Documentation | iVar on GitHub |
| Original Publication(s) | An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar |
QualiMap: BAM File Quality Assessment
QualiMap evaluates the quality of alignment data in BAM files by computing various metrics including coverage distribution, mapping quality, GC content, and various metrics analyzed across the reference. It provides comprehensive quality control reports for next-generation sequencing alignment data.
This task generates both standard QualiMap reports and custom interactive HTML visualizations for genome coverage and mapping quality across the reference sequence. The results are bundled into a compressed archive for easy download and review.
QualiMap Technical Details
| Links | |
|---|---|
| Task | task_qualimap.wdl |
| Software Source Code | QualiMap on Bitbucket |
| Software Documentation | QualiMap Documentation |
| Original Publication(s) | QualiMap: evaluating next-generation sequencing alignment data |
qc_check: Check QC Metrics Against User-Defined Thresholds (optional)
To activate this task, provide a qc_check_table as input.
The QC Check task compares generated QC metrics against user-defined thresholds for each metric. This task will run if the user provides a qc_check_table TSV file. If all QC metrics meet the threshold, the qc_check output variable will read QC_PASS. Otherwise, the output will read QC_NA if the task could not proceed or QC_ALERT followed by a string indicating what metric failed.
Thresholds for percent human classified reads and percent human classified dehosted reads are noted as "classified_human" and "classified_human_dehosted" respectively. In other workflows, these QC check columns are named "kraken_human" or "metabuli_human", but Freyja uses the "classified_" prefix because the classification software used will vary depending on read type (kraken2 for Illumina, Metabuli for ONT).
Formatting the qc_check_table.tsv
- The first column of the qc_check_table lists the
organismthat the task will assess and the header of this column must be "taxon". - Each subsequent column indicates a QC metric and lists a threshold for each organism that will be checked. The column names must exactly match expected values, so we highly recommend copy and pasting the header from the template file below as a starting place.
Template qc_check_table.tsv files
- Freyja_FASTQ: freyja_qc_check_template.tsv
Example Purposes Only
The QC threshold values shown in the file above are for example purposes only and should not be presumed to be sufficient for every dataset.
qc_check Technical Details
| Links | |
|---|---|
| Task | task_qc_check_phb.wdl |
read_QC_trim_ont: Read Quality Trimming, Quantification, and Identification
read_QC_trim_ont is a sub-workflow that filters low-quality reads and trims low-quality regions of reads. It uses several tasks, described below.
HRRT: Human Host Sequence Removal
All reads of human origin are removed, including their mates, by using NCBI's human read removal tool (HRRT).
HRRT is based on the SRA Taxonomy Analysis Tool and employs a k-mer database constructed of k-mers from Eukaryota derived from all human RefSeq records with any k-mers found in non-Eukaryota RefSeq records subtracted from the database.
HRRT Technical Details
| Links | |
|---|---|
| Task | task_ncbi_scrub.wdl |
| Software Source Code | HRRT on GitHub |
| Software Documentation | HRRT on NCBI |
artic guppyplex: Read Filtering
Reads are filtered by length with the artic guppyplex command, which is a part of the ARTIC protocol. Since TheiaCoV was developed primarily for amplicon-based viral sequencing, this task is included to remove chimeric reads that are either too short or too long.
ARTIC guppyplex Technical Details
| Links | |
|---|---|
| Task | task_artic_guppyplex.wdl |
| Software Source Code | ARTIC on GitHub |
| Software Documentation | ARTIC Documentation |
Metabuli: Read Classification
Metabuli is used to classify and optionally extract reads against a reference database. Metabuli uses a novel k-mer structure, called metamer, to analyze both amino acid (AA) and DNA sequences. It leverages AA conservation for sensitive homology detection and DNA mutations for specific differentiation between closely related taxa.
Metabuli is run on both raw and human dehosted reads.
taxdump_path input parameter
The taxdump_path directs the task toward a taxonkit-generated taxdump file, e.g. from NCBI or from GTDB. This is not necessary to edit unless users want a more recent taxdump than what Theiagen hosts, or if users want to reference a different taxonomy. By default, Theiagen uses the NCBI taxonomy hierarchy.
cpu / memory input parameters
Increasing the memory and cpus allocated to Metabuli can substantially increase throughput.
extract_unclassified input parameter
This parameter determines whether unclassified reads should also be extracted and combined with the taxon-specific extracted reads. By default, this is set to false, meaning that only reads classified to the specified input taxon will be extracted.
Metabuli Technical Details
| Links | |
|---|---|
| Task | task_metabuli.wdl |
| Software Source Code | Metabuli on GitHub |
| Software Documentation | Metabuli Documentation |
| Original Publication(s) | Metabuli: sensitive and specific metagenomic classification via joint analysis of amino acid and DNA |
NanoPlot: Read Quantification
NanoPlot is used for the determination of mean quality scores, read lengths, and number of reads. This task is run once with raw reads as input and once with clean reads as input. If QC has been performed correctly, you should expect fewer clean reads than raw reads.
While this task currently is run outside of the read_QC_trim_ont workflow, it is being included here as it calculates statistics on the read data. This is done so that the actual assembly genome lengths can be used (if an estimated genome length is not provided by the user) to ensure the estimated coverage statistics are accurate.
NanoPlot Technical Details
| Links | |
|---|---|
| Task | task_nanoplot.wdl |
| Software Source Code | NanoPlot on GitHub |
| Software Documentation | NanoPlot Documentation |
| Original Publication(s) | NanoPack2: population-scale evaluation of long-read sequencing data |
read_QC_trim_ont Technical Details
| Links | |
|---|---|
| Subworkflow | wf_read_QC_trim_ont.wdl |
Minimap2: Read Alignment
Minimap2 is a popular aligner that is used to align reads (or assemblies) to an assembly file. In Minimap2, "modes" are a group of preset options.
The mode used in this task is map-ont which is the default mode for long reads and indicates that long reads of ~10% error rates should be aligned to the reference genome. The output file is in SAM format.
For more information regarding modes and the available options for Minimap2, please see the Minimap2 manpage.
Minimap2 Technical Details
| Links | |
|---|---|
| Task | task_minimap2.wdl |
| Software Source Code | Minimap2 on GitHub |
| Software Documentation | Minimap2 |
| Original Publication(s) | Minimap2: pairwise alignment for nucleotide sequences |
QualiMap: BAM File Quality Assessment
QualiMap evaluates the quality of alignment data in BAM files by computing various metrics including coverage distribution, mapping quality, GC content, and various metrics analyzed across the reference. It provides comprehensive quality control reports for next-generation sequencing alignment data.
This task generates both standard QualiMap reports and custom interactive HTML visualizations for genome coverage and mapping quality across the reference sequence. The results are bundled into a compressed archive for easy download and review.
QualiMap Technical Details
| Links | |
|---|---|
| Task | task_qualimap.wdl |
| Software Source Code | QualiMap on Bitbucket |
| Software Documentation | QualiMap Documentation |
| Original Publication(s) | QualiMap: evaluating next-generation sequencing alignment data |
qc_check: Check QC Metrics Against User-Defined Thresholds (optional)
To activate this task, provide a qc_check_table as input.
The QC Check task compares generated QC metrics against user-defined thresholds for each metric. This task will run if the user provides a qc_check_table TSV file. If all QC metrics meet the threshold, the qc_check output variable will read QC_PASS. Otherwise, the output will read QC_NA if the task could not proceed or QC_ALERT followed by a string indicating what metric failed.
Thresholds for percent human classified reads and percent human classified dehosted reads are noted as "classified_human" and "classified_human_dehosted" respectively. In other workflows, these QC check columns are named "kraken_human" or "metabuli_human", but Freyja uses the "classified_" prefix because the classification software used will vary depending on read type (kraken2 for Illumina, Metabuli for ONT).
Formatting the qc_check_table.tsv
- The first column of the qc_check_table lists the
organismthat the task will assess and the header of this column must be "taxon". - Each subsequent column indicates a QC metric and lists a threshold for each organism that will be checked. The column names must exactly match expected values, so we highly recommend copy and pasting the header from the template file below as a starting place.
Template qc_check_table.tsv files
- Freyja_FASTQ: freyja_qc_check_template.tsv
Example Purposes Only
The QC threshold values shown in the file above are for example purposes only and should not be presumed to be sufficient for every dataset.
qc_check Technical Details
| Links | |
|---|---|
| Task | task_qc_check_phb.wdl |
gene_coverage: Depth and Breadth of Coverage Calculations
This task calculates average read depth and the percent of a region (typically genes) covered above a minimum depth and quality using samtools (Pysam) and basic arithmetic.
Outputs are reported as JSON-based "maps" that relate gene names to their breadth and depth of coverage. For example:
percent_coverage_by_gene:
depth_by_gene:
By default, this task runs for SARS-CoV-2 and Mpox.
Region coordinates must be relevant to the reference genome
Please note that default BEDfiles contain gene coordinates that may not directly match user-provided or dynamically-selected reference genomes (TheiaViral).
BED file usage
In viral characterization workflows, gene coverage regions are supplied with a BED file.
- To extract custom regions of interest, populate the
reference_gene_locations_bedinput (tasktheiacov/morgana_magic) - If no custom BED is provided, organism defaults are used when available
- BED files should include a gene name in column 4 to label output
Gene Coverage Technical Details
| Links | |
|---|---|
| Task | task_gene_coverage.wdl |
| Software Source Code | samtools on GitHub |
| Software Documentation | samtools Manual |
| Original Publication(s) | Twelve years of SAMtools and BCFtools |
freyja Details
The Freyja task will call variants and capture sequencing depth information to identify the relative abundance of lineages present. Optionally, if bootstrap is set to true, bootstrapping will be performed. After the optional bootstrapping step, the variants are demixed.
Freyja Technical Details
| Links | |
|---|---|
| Task | task_freyja.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://andersen-lab.github.io/Freyja/index.html# |
freyja_long_format Details
The freyja_long_format task converts the demixed lineage abundances for a single sample into a long-format TSV that is paired with the sample's metadata (collection date, collection site, genome coverage, and optionally latitude and longitude). Collection site, collection date, and genome coverage are necessary inputs to produce the long format. This long-format TSV is suitable for downstream aggregation across samples and for use with visualization tools such as Microreact.
The sample's genome coverage (freyja.freyja_coverage) is included automatically so that the --mincov threshold can drop the sample from the output when its coverage falls below freyja_min_coverage.
Lineage grouping can be customized by providing the optional group_by input, which will group by collection site + collection date, or by collection site + week and normalize the data.
Behavior when the sample fails the coverage threshold
The minimum genome coverage threshold is controlled by the freyja_min_coverage workflow input (default: 60) and is passed to task. If the sample's freyja_coverage falls below this threshold, no lineage rows are written and the resulting freyja_parsed_format_tsv instead contains the text all samples are below coverage. Lower the freyja_min_coverage input if you wish to retain low-coverage samples in downstream visualizations.
Freyja Long Format Technical Details
| Links | |
|---|---|
| Task | task_freyja_long_way.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://andersen-lab.github.io/Freyja/index.html# |
Outputs¶
The main output file used in subsequent Freyja workflows is found under the freyja_demixed column. This TSV file takes on the following format:
| sample name | |
|---|---|
| summarized | [('Delta', 0.65), ('Other', 0.25), ('Alpha', 0.1)] |
| lineages | ['B.1.617.2' 'B.1.2' 'AY.6' 'Q.3'] |
| abundances | "[0.5 0.25 0.15 0.1]" |
| resid | 3.14159 |
| coverage | 95.8 |
- The
summarizedarray denotes a sum of all lineage abundances in a particular WHO designation (i.e. B.1.617.2 and AY.6 abundances are summed in the above example), otherwise they are grouped into "Other". - The
lineagearray lists the identified lineages in descending order - The
abundancesarray contains the corresponding abundance estimates. - The value of
residcorresponds to the residual of the weighted least absolute deviation problem used to estimate lineage abundances. - The
coveragevalue provides the 10x coverage estimate (percent of sites with 10 or greater reads)
Click 'Ignore empty outputs'
When running the Freyja_FASTQ_PHB workflow, it is recommended to select the "Ignore empty outputs" option in the Terra UI. This will hide the output columns that will not be generated for your input data type.
| Variable | Type | Description |
|---|---|---|
| aligned_bai | String | Index companion file to the bam file generated during the consensus assembly process |
| aligned_bam | String | Sorted BAM file containing the alignments of reads to the reference genome |
| alignment_method | String | The method used to generate the alignment |
| bbduk_docker | String | The Docker image for bbduk, which was used to remove the adapters from the sequences |
| bracken_report | String | Refined kraken2 report generated by Bracken |
| bracken_report_dehosted | String | Dehosted refined kraken2 report generated by Bracken |
| bwa_version | String | Version of BWA software used |
| est_percent_gene_coverage_tsv | File | Percent coverage for each gene in the organism being analyzed (depending on the organism input) |
| fastp_docker | String | Docker image used for fastp |
| fastp_html_report | String | The HTML report conveying fastp results |
| fastp_json_report | String | The JSON report conveying fastp results |
| fastp_version | String | The version of fastp used |
| fastq_scan_clean1_json | String | The JSON file output from fastq-scan containing summary stats about clean forward read quality and length |
| fastq_scan_clean2_json | File | The JSON file output from fastq-scan containing summary stats about clean reverse read quality and length |
| fastq_scan_num_reads_clean1 | String | The number of forward reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean2 | Int | The number of reverse reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean_pairs | String | The number of read pairs after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_raw1 | String | The number of input forward reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw2 | Int | The number of input reserve reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw_pairs | String | The number of input read pairs as calculated by fastq_scan |
| fastq_scan_raw1_json | String | The JSON file output from fastq-scan containing summary stats about raw forward read quality and length |
| fastq_scan_raw2_json | File | The JSON file output from fastq-scan containing summary stats about raw reverse read quality and length |
| fastq_scan_version | String | The version of fastq_scan |
| fastqc_clean1_html | String | An HTML file that provides a graphical visualization of clean forward read quality from fastqc to open in an internet browser |
| fastqc_clean2_html | File | An HTML file that provides a graphical visualization of clean reverse read quality from fastqc to open in an internet browser |
| fastqc_docker | String | The Docker container used for fastqc |
| fastqc_num_reads_clean1 | String | The number of forward reads after cleaning by fastqc |
| fastqc_num_reads_clean2 | Int | The number of reverse reads after cleaning by fastqc |
| fastqc_num_reads_clean_pairs | String | The number of read pairs after cleaning by fastqc |
| fastqc_num_reads_raw1 | String | The number of input forward reads by fastqc before cleaning |
| fastqc_num_reads_raw2 | Int | The number of input reverse reads by fastqc before cleaning |
| fastqc_num_reads_raw_pairs | String | The number of input read pairs by fastqc before cleaning |
| fastqc_raw1_html | String | An HTML file that provides a graphical visualization of raw forward read quality from fastqc to open in an internet browser |
| fastqc_raw2_html | File | An HTML file that provides a graphical visualization of raw reverse read quality from fastqc to open in an internet browser |
| fastqc_version | String | Version of fastqc software used |
| freyja_abundances | String | Abundances estimates identified by Freyja and parsed from freyja_demixed file |
| freyja_barcode_file | String | Barcode file used with Freyja |
| freyja_barcode_version | String | Name of barcode file used, or the date if update_db is true |
| freyja_bootstrap_lineages | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each lineage |
| freyja_bootstrap_lineages_pdf | String | A boxplot of the bootstrap lineages CSV file |
| freyja_bootstrap_summary | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each WHO designated VOI/VOC |
| freyja_bootstrap_summary_pdf | String | A boxplot of the bootstrap summary CSV file |
| freyja_coverage | Float | Coverage identified by Freyja and parsed from freyja_demixed file |
| freyja_demixed | File | The main output TSV; see the section directly above this table for an explanation |
| freyja_demixed_parsed | File | Parsed freyja_demixed file, containing the same information, for easy result concatenation |
| freyja_depths | File | A TSV listing the depth of every position |
| freyja_fastq_wf_analysis_date | String | Date of analysis |
| freyja_fastq_wf_version | String | The version of the Public Health Bioinformatics (PHB) repository used |
| freyja_lineage_metadata_file | String | Metadata file for lineages identified by Freyja |
| freyja_lineages | String | Lineages in descending order identified by Freyja and parsed from freyja_demixed file |
| freyja_long_format_docker_used | String | Docker image used |
| freyja_metadata_version | String | Name of lineage metadata file used, or the date if update_db is true |
| freyja_parsed_format_tsv | File | Long-format TSV pairing freyja lineage abundances with sample metadata (collection date, collection site, latitude, longitude); produced by the freyja_long_format task and consumed by freyja_microreact |
| freyja_resid | String | Residual of the weighted least absolute deviation problem used to estimate lineage abundances identified by Freyja and parsed from freyja_demixed file |
| freyja_summarized | String | Sum of all lineage abundances in a particular WHO designation identified by Freyja and parsed from freyja_demixed file |
| freyja_variants | File | The TSV file containing the variants identified by Freyja |
| freyja_version | String | version of Freyja used |
| gene_coverage_depth_by_gene | Map[String,Float] | A JSON map relating gene queries to their depth of coverage |
| gene_coverage_percent_coverage_by_gene | Map[String,Float] | A JSON map relating gene queries to their percent breadth of coverage |
| ivar_version_primtrim | String | Version of iVar for running the iVar trim command |
| kraken_human | String | Percent of human read data detected using the Kraken2 software |
| kraken_human_dehosted | String | Percent of human read data detected using the Kraken2 software after host removal |
| kraken_report | String | Full Kraken report |
| kraken_report_dehosted | String | Full Kraken report after host removal |
| kraken_version | String | Version of Kraken software used |
| metabuli_human | Float | Percent of human reads detected in raw reads |
| metabuli_human_dehosted | Float | Percent of human reads detected after removing human reads |
| metabuli_report | String | Classification report from Metabuli |
| metabuli_report_dehosted | String | Classification report from Metabuli after removing human reads |
| metabuli_version | String | Version of Metabuli used |
| minimap2_docker | String | The Docker image of minimap2 |
| minimap2_version | String | The version of minimap2 |
| nanoplot_html_clean | File | An HTML report describing the clean reads |
| nanoplot_html_raw | File | An HTML report describing the raw reads |
| nanoplot_num_reads_clean1 | Int | Number of clean reads |
| nanoplot_num_reads_raw1 | Int | Number of raw reads |
| nanoplot_r1_est_coverage_clean | Float | Estimated coverage on the clean reads by nanoplot |
| nanoplot_r1_est_coverage_raw | Float | Estimated coverage on the raw reads by nanoplot |
| nanoplot_r1_mean_q_clean | Float | Mean quality score of clean forward reads |
| nanoplot_r1_mean_q_raw | Float | Mean quality score of raw forward reads |
| nanoplot_r1_mean_readlength_clean | Float | Mean read length of clean forward reads |
| nanoplot_r1_mean_readlength_raw | Float | Mean read length of raw forward reads |
| nanoplot_r1_median_q_clean | Float | Median quality score of clean forward reads |
| nanoplot_r1_median_q_raw | Float | Median quality score of raw forward reads |
| nanoplot_r1_median_readlength_clean | Float | Median read length of clean forward reads |
| nanoplot_r1_median_readlength_raw | Float | Median read length of raw forward reads |
| nanoplot_r1_n50_clean | Float | N50 of clean forward reads |
| nanoplot_r1_n50_raw | Float | N50 of raw forward reads |
| nanoplot_r1_stdev_readlength_clean | Float | Standard deviation read length of clean forward reads |
| nanoplot_r1_stdev_readlength_raw | Float | Standard deviation read length of raw forward reads |
| nanoplot_tsv_clean | File | A TSV report describing the clean reads |
| nanoplot_tsv_raw | File | A TSV report describing the raw reads |
| nanoq_version | String | Version of nanoq used in analysis |
| primer_bed_name | String | Name of the primer bed files used for primer trimming |
| primer_trimmed_read_percent | Float | Percentage of read data with primers trimmed as determined by iVar trim |
| qc_check | String | A string that indicates whether or not the sample passes a set of pre-determined and user-provided QC thresholds |
| qc_standard | File | The file used in the QC Check task containing the QC thresholds. |
| qualimap_coverage_plots_html | File | Interactive HTML Plots of Coverage Across the Genome |
| qualimap_docker | String | Qualimap docker image used |
| qualimap_reports_bundle | File | Zipped bundle of Qualimap reports and plots |
| qualimap_version | String | Version of Qualimap used |
| read1_clean | File | Forward read file after quality trimming and adapter removal |
| read1_dehosted | File | The dehosted forward reads file; suggested read file for SRA submission |
| read2_clean | File | Reverse read file after quality trimming and adapter removal |
| read2_dehosted | File | The dehosted reverse reads file; suggested read file for SRA submission |
| samtools_version | String | The version of samtools used to sort and index the alignment file |
| samtools_version_primtrim | String | The version of samtools used to create the pileup before running iVar trim |
| sc2_s_gene_mean_coverage | Float | Mean read depth for the S gene in SARS-CoV-2 |
| sc2_s_gene_percent_coverage | Float | Percent coverage of the S gene in SARS-CoV-2 |
| trimmomatic_docker | String | The docker image used for the trimmomatic module in this workflow |
| trimmomatic_version | String | The version of Trimmomatic used |
| Variable | Type | Description |
|---|---|---|
| aligned_bai | String | Index companion file to the bam file generated during the consensus assembly process |
| aligned_bam | String | Sorted BAM file containing the alignments of reads to the reference genome |
| alignment_method | String | The method used to generate the alignment |
| bbduk_docker | String | The Docker image for bbduk, which was used to remove the adapters from the sequences |
| bracken_report | String | Refined kraken2 report generated by Bracken |
| bracken_report_dehosted | String | Dehosted refined kraken2 report generated by Bracken |
| bwa_version | String | Version of BWA software used |
| est_percent_gene_coverage_tsv | File | Percent coverage for each gene in the organism being analyzed (depending on the organism input) |
| fastp_docker | String | Docker image used for fastp |
| fastp_html_report | String | The HTML report conveying fastp results |
| fastp_json_report | String | The JSON report conveying fastp results |
| fastp_version | String | The version of fastp used |
| fastq_scan_clean1_json | String | The JSON file output from fastq-scan containing summary stats about clean forward read quality and length |
| fastq_scan_clean2_json | File | The JSON file output from fastq-scan containing summary stats about clean reverse read quality and length |
| fastq_scan_num_reads_clean1 | String | The number of forward reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean2 | Int | The number of reverse reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean_pairs | String | The number of read pairs after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_raw1 | String | The number of input forward reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw2 | Int | The number of input reserve reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw_pairs | String | The number of input read pairs as calculated by fastq_scan |
| fastq_scan_raw1_json | String | The JSON file output from fastq-scan containing summary stats about raw forward read quality and length |
| fastq_scan_raw2_json | File | The JSON file output from fastq-scan containing summary stats about raw reverse read quality and length |
| fastq_scan_version | String | The version of fastq_scan |
| fastqc_clean1_html | String | An HTML file that provides a graphical visualization of clean forward read quality from fastqc to open in an internet browser |
| fastqc_clean2_html | File | An HTML file that provides a graphical visualization of clean reverse read quality from fastqc to open in an internet browser |
| fastqc_docker | String | The Docker container used for fastqc |
| fastqc_num_reads_clean1 | String | The number of forward reads after cleaning by fastqc |
| fastqc_num_reads_clean2 | Int | The number of reverse reads after cleaning by fastqc |
| fastqc_num_reads_clean_pairs | String | The number of read pairs after cleaning by fastqc |
| fastqc_num_reads_raw1 | String | The number of input forward reads by fastqc before cleaning |
| fastqc_num_reads_raw2 | Int | The number of input reverse reads by fastqc before cleaning |
| fastqc_num_reads_raw_pairs | String | The number of input read pairs by fastqc before cleaning |
| fastqc_raw1_html | String | An HTML file that provides a graphical visualization of raw forward read quality from fastqc to open in an internet browser |
| fastqc_raw2_html | File | An HTML file that provides a graphical visualization of raw reverse read quality from fastqc to open in an internet browser |
| fastqc_version | String | Version of fastqc software used |
| freyja_abundances | String | Abundances estimates identified by Freyja and parsed from freyja_demixed file |
| freyja_barcode_file | String | Barcode file used with Freyja |
| freyja_barcode_version | String | Name of barcode file used, or the date if update_db is true |
| freyja_bootstrap_lineages | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each lineage |
| freyja_bootstrap_lineages_pdf | String | A boxplot of the bootstrap lineages CSV file |
| freyja_bootstrap_summary | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each WHO designated VOI/VOC |
| freyja_bootstrap_summary_pdf | String | A boxplot of the bootstrap summary CSV file |
| freyja_coverage | Float | Coverage identified by Freyja and parsed from freyja_demixed file |
| freyja_demixed | File | The main output TSV; see the section directly above this table for an explanation |
| freyja_demixed_parsed | File | Parsed freyja_demixed file, containing the same information, for easy result concatenation |
| freyja_depths | File | A TSV listing the depth of every position |
| freyja_fastq_wf_analysis_date | String | Date of analysis |
| freyja_fastq_wf_version | String | The version of the Public Health Bioinformatics (PHB) repository used |
| freyja_lineage_metadata_file | String | Metadata file for lineages identified by Freyja |
| freyja_lineages | String | Lineages in descending order identified by Freyja and parsed from freyja_demixed file |
| freyja_long_format_docker_used | String | Docker image used |
| freyja_metadata_version | String | Name of lineage metadata file used, or the date if update_db is true |
| freyja_parsed_format_tsv | File | Long-format TSV pairing freyja lineage abundances with sample metadata (collection date, collection site, latitude, longitude); produced by the freyja_long_format task and consumed by freyja_microreact |
| freyja_resid | String | Residual of the weighted least absolute deviation problem used to estimate lineage abundances identified by Freyja and parsed from freyja_demixed file |
| freyja_summarized | String | Sum of all lineage abundances in a particular WHO designation identified by Freyja and parsed from freyja_demixed file |
| freyja_variants | File | The TSV file containing the variants identified by Freyja |
| freyja_version | String | version of Freyja used |
| gene_coverage_depth_by_gene | Map[String,Float] | A JSON map relating gene queries to their depth of coverage |
| gene_coverage_percent_coverage_by_gene | Map[String,Float] | A JSON map relating gene queries to their percent breadth of coverage |
| ivar_version_primtrim | String | Version of iVar for running the iVar trim command |
| kraken_human | String | Percent of human read data detected using the Kraken2 software |
| kraken_human_dehosted | String | Percent of human read data detected using the Kraken2 software after host removal |
| kraken_report | String | Full Kraken report |
| kraken_report_dehosted | String | Full Kraken report after host removal |
| kraken_version | String | Version of Kraken software used |
| metabuli_human | Float | Percent of human reads detected in raw reads |
| metabuli_human_dehosted | Float | Percent of human reads detected after removing human reads |
| metabuli_report | String | Classification report from Metabuli |
| metabuli_report_dehosted | String | Classification report from Metabuli after removing human reads |
| metabuli_version | String | Version of Metabuli used |
| minimap2_docker | String | The Docker image of minimap2 |
| minimap2_version | String | The version of minimap2 |
| nanoplot_html_clean | File | An HTML report describing the clean reads |
| nanoplot_html_raw | File | An HTML report describing the raw reads |
| nanoplot_num_reads_clean1 | Int | Number of clean reads |
| nanoplot_num_reads_raw1 | Int | Number of raw reads |
| nanoplot_r1_est_coverage_clean | Float | Estimated coverage on the clean reads by nanoplot |
| nanoplot_r1_est_coverage_raw | Float | Estimated coverage on the raw reads by nanoplot |
| nanoplot_r1_mean_q_clean | Float | Mean quality score of clean forward reads |
| nanoplot_r1_mean_q_raw | Float | Mean quality score of raw forward reads |
| nanoplot_r1_mean_readlength_clean | Float | Mean read length of clean forward reads |
| nanoplot_r1_mean_readlength_raw | Float | Mean read length of raw forward reads |
| nanoplot_r1_median_q_clean | Float | Median quality score of clean forward reads |
| nanoplot_r1_median_q_raw | Float | Median quality score of raw forward reads |
| nanoplot_r1_median_readlength_clean | Float | Median read length of clean forward reads |
| nanoplot_r1_median_readlength_raw | Float | Median read length of raw forward reads |
| nanoplot_r1_n50_clean | Float | N50 of clean forward reads |
| nanoplot_r1_n50_raw | Float | N50 of raw forward reads |
| nanoplot_r1_stdev_readlength_clean | Float | Standard deviation read length of clean forward reads |
| nanoplot_r1_stdev_readlength_raw | Float | Standard deviation read length of raw forward reads |
| nanoplot_tsv_clean | File | A TSV report describing the clean reads |
| nanoplot_tsv_raw | File | A TSV report describing the raw reads |
| nanoq_version | String | Version of nanoq used in analysis |
| primer_bed_name | String | Name of the primer bed files used for primer trimming |
| primer_trimmed_read_percent | Float | Percentage of read data with primers trimmed as determined by iVar trim |
| qc_check | String | A string that indicates whether or not the sample passes a set of pre-determined and user-provided QC thresholds |
| qc_standard | File | The file used in the QC Check task containing the QC thresholds. |
| qualimap_coverage_plots_html | File | Interactive HTML Plots of Coverage Across the Genome |
| qualimap_docker | String | Qualimap docker image used |
| qualimap_reports_bundle | File | Zipped bundle of Qualimap reports and plots |
| qualimap_version | String | Version of Qualimap used |
| read1_clean | File | Forward read file after quality trimming and adapter removal |
| read1_dehosted | File | The dehosted forward reads file; suggested read file for SRA submission |
| read2_clean | File | Reverse read file after quality trimming and adapter removal |
| read2_dehosted | File | The dehosted reverse reads file; suggested read file for SRA submission |
| samtools_version | String | The version of samtools used to sort and index the alignment file |
| samtools_version_primtrim | String | The version of samtools used to create the pileup before running iVar trim |
| sc2_s_gene_mean_coverage | Float | Mean read depth for the S gene in SARS-CoV-2 |
| sc2_s_gene_percent_coverage | Float | Percent coverage of the S gene in SARS-CoV-2 |
| trimmomatic_docker | String | The docker image used for the trimmomatic module in this workflow |
| trimmomatic_version | String | The version of Trimmomatic used |
| Variable | Type | Description |
|---|---|---|
| aligned_bai | String | Index companion file to the bam file generated during the consensus assembly process |
| aligned_bam | String | Sorted BAM file containing the alignments of reads to the reference genome |
| alignment_method | String | The method used to generate the alignment |
| bbduk_docker | String | The Docker image for bbduk, which was used to remove the adapters from the sequences |
| bracken_report | String | Refined kraken2 report generated by Bracken |
| bracken_report_dehosted | String | Dehosted refined kraken2 report generated by Bracken |
| bwa_version | String | Version of BWA software used |
| est_percent_gene_coverage_tsv | File | Percent coverage for each gene in the organism being analyzed (depending on the organism input) |
| fastp_docker | String | Docker image used for fastp |
| fastp_html_report | String | The HTML report conveying fastp results |
| fastp_json_report | String | The JSON report conveying fastp results |
| fastp_version | String | The version of fastp used |
| fastq_scan_clean1_json | String | The JSON file output from fastq-scan containing summary stats about clean forward read quality and length |
| fastq_scan_clean2_json | File | The JSON file output from fastq-scan containing summary stats about clean reverse read quality and length |
| fastq_scan_num_reads_clean1 | String | The number of forward reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean2 | Int | The number of reverse reads after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_clean_pairs | String | The number of read pairs after cleaning as calculated by fastq_scan |
| fastq_scan_num_reads_raw1 | String | The number of input forward reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw2 | Int | The number of input reserve reads as calculated by fastq_scan |
| fastq_scan_num_reads_raw_pairs | String | The number of input read pairs as calculated by fastq_scan |
| fastq_scan_raw1_json | String | The JSON file output from fastq-scan containing summary stats about raw forward read quality and length |
| fastq_scan_raw2_json | File | The JSON file output from fastq-scan containing summary stats about raw reverse read quality and length |
| fastq_scan_version | String | The version of fastq_scan |
| fastqc_clean1_html | String | An HTML file that provides a graphical visualization of clean forward read quality from fastqc to open in an internet browser |
| fastqc_clean2_html | File | An HTML file that provides a graphical visualization of clean reverse read quality from fastqc to open in an internet browser |
| fastqc_docker | String | The Docker container used for fastqc |
| fastqc_num_reads_clean1 | String | The number of forward reads after cleaning by fastqc |
| fastqc_num_reads_clean2 | Int | The number of reverse reads after cleaning by fastqc |
| fastqc_num_reads_clean_pairs | String | The number of read pairs after cleaning by fastqc |
| fastqc_num_reads_raw1 | String | The number of input forward reads by fastqc before cleaning |
| fastqc_num_reads_raw2 | Int | The number of input reverse reads by fastqc before cleaning |
| fastqc_num_reads_raw_pairs | String | The number of input read pairs by fastqc before cleaning |
| fastqc_raw1_html | String | An HTML file that provides a graphical visualization of raw forward read quality from fastqc to open in an internet browser |
| fastqc_raw2_html | File | An HTML file that provides a graphical visualization of raw reverse read quality from fastqc to open in an internet browser |
| fastqc_version | String | Version of fastqc software used |
| freyja_abundances | String | Abundances estimates identified by Freyja and parsed from freyja_demixed file |
| freyja_barcode_file | String | Barcode file used with Freyja |
| freyja_barcode_version | String | Name of barcode file used, or the date if update_db is true |
| freyja_bootstrap_lineages | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each lineage |
| freyja_bootstrap_lineages_pdf | String | A boxplot of the bootstrap lineages CSV file |
| freyja_bootstrap_summary | String | A CSV that contains the 0.025, 0.05, 0.25, 0.5 (median), 0.75, 0.95, and 0.975 percentiles for each WHO designated VOI/VOC |
| freyja_bootstrap_summary_pdf | String | A boxplot of the bootstrap summary CSV file |
| freyja_coverage | Float | Coverage identified by Freyja and parsed from freyja_demixed file |
| freyja_demixed | File | The main output TSV; see the section directly above this table for an explanation |
| freyja_demixed_parsed | File | Parsed freyja_demixed file, containing the same information, for easy result concatenation |
| freyja_depths | File | A TSV listing the depth of every position |
| freyja_fastq_wf_analysis_date | String | Date of analysis |
| freyja_fastq_wf_version | String | The version of the Public Health Bioinformatics (PHB) repository used |
| freyja_lineage_metadata_file | String | Metadata file for lineages identified by Freyja |
| freyja_lineages | String | Lineages in descending order identified by Freyja and parsed from freyja_demixed file |
| freyja_long_format_docker_used | String | Docker image used |
| freyja_metadata_version | String | Name of lineage metadata file used, or the date if update_db is true |
| freyja_parsed_format_tsv | File | Long-format TSV pairing freyja lineage abundances with sample metadata (collection date, collection site, latitude, longitude); produced by the freyja_long_format task and consumed by freyja_microreact |
| freyja_resid | String | Residual of the weighted least absolute deviation problem used to estimate lineage abundances identified by Freyja and parsed from freyja_demixed file |
| freyja_summarized | String | Sum of all lineage abundances in a particular WHO designation identified by Freyja and parsed from freyja_demixed file |
| freyja_variants | File | The TSV file containing the variants identified by Freyja |
| freyja_version | String | version of Freyja used |
| gene_coverage_depth_by_gene | Map[String,Float] | A JSON map relating gene queries to their depth of coverage |
| gene_coverage_percent_coverage_by_gene | Map[String,Float] | A JSON map relating gene queries to their percent breadth of coverage |
| ivar_version_primtrim | String | Version of iVar for running the iVar trim command |
| kraken_human | String | Percent of human read data detected using the Kraken2 software |
| kraken_human_dehosted | String | Percent of human read data detected using the Kraken2 software after host removal |
| kraken_report | String | Full Kraken report |
| kraken_report_dehosted | String | Full Kraken report after host removal |
| kraken_version | String | Version of Kraken software used |
| metabuli_human | Float | Percent of human reads detected in raw reads |
| metabuli_human_dehosted | Float | Percent of human reads detected after removing human reads |
| metabuli_report | String | Classification report from Metabuli |
| metabuli_report_dehosted | String | Classification report from Metabuli after removing human reads |
| metabuli_version | String | Version of Metabuli used |
| minimap2_docker | String | The Docker image of minimap2 |
| minimap2_version | String | The version of minimap2 |
| nanoplot_html_clean | File | An HTML report describing the clean reads |
| nanoplot_html_raw | File | An HTML report describing the raw reads |
| nanoplot_num_reads_clean1 | Int | Number of clean reads |
| nanoplot_num_reads_raw1 | Int | Number of raw reads |
| nanoplot_r1_est_coverage_clean | Float | Estimated coverage on the clean reads by nanoplot |
| nanoplot_r1_est_coverage_raw | Float | Estimated coverage on the raw reads by nanoplot |
| nanoplot_r1_mean_q_clean | Float | Mean quality score of clean forward reads |
| nanoplot_r1_mean_q_raw | Float | Mean quality score of raw forward reads |
| nanoplot_r1_mean_readlength_clean | Float | Mean read length of clean forward reads |
| nanoplot_r1_mean_readlength_raw | Float | Mean read length of raw forward reads |
| nanoplot_r1_median_q_clean | Float | Median quality score of clean forward reads |
| nanoplot_r1_median_q_raw | Float | Median quality score of raw forward reads |
| nanoplot_r1_median_readlength_clean | Float | Median read length of clean forward reads |
| nanoplot_r1_median_readlength_raw | Float | Median read length of raw forward reads |
| nanoplot_r1_n50_clean | Float | N50 of clean forward reads |
| nanoplot_r1_n50_raw | Float | N50 of raw forward reads |
| nanoplot_r1_stdev_readlength_clean | Float | Standard deviation read length of clean forward reads |
| nanoplot_r1_stdev_readlength_raw | Float | Standard deviation read length of raw forward reads |
| nanoplot_tsv_clean | File | A TSV report describing the clean reads |
| nanoplot_tsv_raw | File | A TSV report describing the raw reads |
| nanoq_version | String | Version of nanoq used in analysis |
| primer_bed_name | String | Name of the primer bed files used for primer trimming |
| primer_trimmed_read_percent | Float | Percentage of read data with primers trimmed as determined by iVar trim |
| qc_check | String | A string that indicates whether or not the sample passes a set of pre-determined and user-provided QC thresholds |
| qc_standard | File | The file used in the QC Check task containing the QC thresholds. |
| qualimap_coverage_plots_html | File | Interactive HTML Plots of Coverage Across the Genome |
| qualimap_docker | String | Qualimap docker image used |
| qualimap_reports_bundle | File | Zipped bundle of Qualimap reports and plots |
| qualimap_version | String | Version of Qualimap used |
| read1_clean | File | Forward read file after quality trimming and adapter removal |
| read1_dehosted | File | The dehosted forward reads file; suggested read file for SRA submission |
| read2_clean | File | Reverse read file after quality trimming and adapter removal |
| read2_dehosted | File | The dehosted reverse reads file; suggested read file for SRA submission |
| samtools_version | String | The version of samtools used to sort and index the alignment file |
| samtools_version_primtrim | String | The version of samtools used to create the pileup before running iVar trim |
| sc2_s_gene_mean_coverage | Float | Mean read depth for the S gene in SARS-CoV-2 |
| sc2_s_gene_percent_coverage | Float | Percent coverage of the S gene in SARS-CoV-2 |
| trimmomatic_docker | String | The docker image used for the trimmomatic module in this workflow |
| trimmomatic_version | String | The version of Trimmomatic used |
Freyja_Plot_PHB¶
This workflow visualizes aggregated freyja_demixed output files produced by Freyja_FASTQ_PHB in a single plot (pdf format) which provides fractional abundance estimates for all aggregated samples.
Options exist to provide lineage-specific breakdowns and/or sample collection time information.
In addition to the aggregate plot, Freyja_Plot_PHB can produce a long-format metadata TSV (freyja_parsed_format_tsv) that combines lineage abundances with per-sample metadata (collection date, collection site, latitude, longitude), as well as a Microreact-compatible upload file (freyja_microreact_output) for interactive geospatial and temporal visualization of the aggregated results.
Inputs¶
This workflow runs on the set level.
| Terra Task Name | Variable | Type | Description | Default Value | Terra Status |
|---|---|---|---|---|---|
| freyja_plot | freyja_demixed | Array[File] | An array containing the output files (freyja_demixed) made by Freyja_FASTQ | Required | |
| freyja_plot | freyja_plot_name | String | The name of the plot to be produced. Example: "my-freyja-plot" | Required | |
| freyja_plot | samplename | Array[String] | The names of the samples being analyzed | Required | |
| freyja_long_format | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja_long_format | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| freyja_long_format | group_by | String | Whether to group samples by collection date or week, options are "date" or "week" | Optional | |
| freyja_long_format | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 4 | Optional |
| freyja_microreact | cpu | Int | Number of CPUs to allocate to the task | 2 | Optional |
| freyja_microreact | disk_size | Int | Amount of storage (in GB) to allocate to the task | 50 | Optional |
| freyja_microreact | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 4 | Optional |
| freyja_plot | collection_date | Array[String] | An array containing the collection dates for the sample (YYYY-MM-DD format) | Optional | |
| freyja_plot | collection_site | Array[String] | An array containing the collection sites for the sample | Optional | |
| freyja_plot | freyja_abundances | Array[String] | An array containing the Freyja abundances for the sample | Optional | |
| freyja_plot | freyja_coverages | Array[Float] | An array containing the genome coverage value (freyja_coverage) for each sample; required to enable the freyja_long_format and freyja_microreact tasks and used by --mincov filtering | Optional | |
| freyja_plot | freyja_lineages | Array[String] | An array containing the Freyja lineages for the sample | Optional | |
| freyja_plot | freyja_long_format_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/freyja-microreact:1.0.2 | Optional |
| freyja_plot | freyja_microreact_docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/freyja-microreact:1.0.2 | Optional |
| freyja_plot | freyja_min_coverage | Int | Minimum genome coverage threshold; passed as --mincov to freyja_to_long.py and as mincov to the freyja plot task | 60 | Optional |
| freyja_plot | latitude | Array[Float] | An array containing the latitudes for the sample collection sites | Optional | |
| freyja_plot | longitude | Array[Float] | An array containing the longitudes for the sample collection sites | Optional | |
| freyja_plot_task | cpu | Int | Number of CPUs to allocate to the task | 1 | Optional |
| freyja_plot_task | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| freyja_plot_task | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/freyja:2.0.1 | Optional |
| freyja_plot_task | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| freyja_plot_task | plot_day_window | Int | The width of the rolling average window; only used if plot_time_interval is "D" | 14 | Optional |
| freyja_plot_task | plot_lineages | Boolean | If true, will plot a lineage-specific breakdown | False | Optional |
| freyja_plot_task | plot_time | Boolean | If true, will plot sample collection time information (requires the collection_date input variable) | False | Optional |
| freyja_plot_task | plot_time_interval | String | Options: "MS" for month, "D" for day | MS | Optional |
| version_capture | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/alpine-plus-bash:3.20.0 | Optional |
| version_capture | timezone | String | Set the time zone to get an accurate date of analysis (uses UTC by default) | Optional |
Analysis Tasks¶
freyja_plot_task Details
This task will aggregate multiple samples together, and then creates a plot. Several optional inputs dictate the plot appearance (see each variable's description for more information).
Freyja Plot Technical Details
| Links | |
|---|---|
| Task | task_freyja_plot.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://github.com/andersen-lab/Freyja |
freyja_long_format Details
The freyja_long_format task aggregates the demixed lineage abundances from multiple samples into a single long-format TSV, paired with each sample's metadata (collection date, collection site, per-sample genome coverage, and optionally latitude and longitude). This long-format TSV is consumed by the freyja_microreact task and is also useful as a standalone input to other visualization or analytical tools.
Per-sample genome coverage values must be supplied via the freyja_coverages array input (typically populated from the freyja_coverage output of Freyja_FASTQ_PHB); this is required so that the --mincov threshold can filter low-coverage samples out of the aggregated TSV.
Lineage grouping can be customized by providing the optional group_by input, which is passed through to the underlying freyja_to_long.py helper script.
Behavior when all samples fail the coverage threshold
The minimum genome coverage threshold is controlled by the freyja_min_coverage workflow input (default: 60) and is passed to freyja_to_long.py as --mincov. Samples whose freyja_coverage falls below this threshold are dropped from the aggregated output. If every sample in the set is below threshold, no lineage rows are written and the resulting freyja_parsed_format_tsv instead contains the sentinel text all samples are below coverage. The downstream freyja_microreact task detects this text and emits an empty freyja_microreact_output file rather than failing — see the freyja_microreact task block below for details. Lower the freyja_min_coverage input if you wish to retain low-coverage samples.
Freyja Long Format Technical Details
| Links | |
|---|---|
| Task | task_freyja_long_way.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://github.com/andersen-lab/Freyja |
freyja_microreact Details
The freyja_microreact task converts the aggregated parsed long-format TSV produced by freyja_long_format into a Microreact-compatible upload file. This output can be uploaded directly to Microreact to interactively explore lineage abundances across samples in time and space. Provide latitude and longitude inputs for geospatial mapping.
Behavior when all samples fail the coverage threshold
Before creating the microreact file, the task inspects the incoming freyja_parsed_format_tsv for the text all samples are below coverage (written upstream by freyja_long_format when no samples passed the freyja_min_coverage threshold). If no samples passed coverage, the task short-circuits and produces an empty freyja_microreact_output file rather than failing the workflow. An empty .microreact file is therefore the expected signal that no samples cleared the coverage threshold; lower the freyja_min_coverage input and rerun if you wish to retain low-coverage samples.
Freyja Microreact Technical Details
| Links | |
|---|---|
| Task | task_freyja_microreact.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://microreact.org/showcase |
Outputs¶
| Variable | Type | Description |
|---|---|---|
| freyja_demixed_aggregate | File | A TSV file that summarizes the freyja_demixed outputs for all samples |
| freyja_long_format_docker_used | String | Docker image used |
| freyja_microreact_docker_used | String | Docker image used |
| freyja_microreact_output | File | Microreact output file for freyja lineages and abundances |
| freyja_parsed_format_tsv | File | Long-format TSV pairing freyja lineage abundances with sample metadata (collection date, collection site, latitude, longitude); produced by the freyja_long_format task and consumed by freyja_microreact |
| freyja_plot | File | A PDF of the plot produced by the workflow |
| freyja_plot_metadata | File | The metadata used to create the plot |
| freyja_plot_version | String | The version of Freyja used |
| freyja_plot_wf_analysis_date | String | The date of analysis |
| freyja_plot_wf_version | String | The version of the Public Health Bioinformatics (PHB) repository used |
Freyja_Dashboard_PHB¶
This workflow creates a group of interactive visualizations based off of the aggregated freyja_demixed output files produced by Freyja_FASTQ_PHB called a "dashboard". Creating this dashboard requires knowing the viral load of your samples (viral copies/liter).
Warning
This dashboard is not "live" — that is, you must rerun the workflow every time you want new data to be included in the visualizations.
Inputs¶
This workflow runs on the set level.
| Terra Task Name | Variable | Type | Description | Default Value | Terra Status |
|---|---|---|---|---|---|
| freyja_dashboard | collection_date | Array[String] | An array containing the collection dates for the sample (YYYY-MM-DD format) | Required | |
| freyja_dashboard | freyja_dashboard_title | String | The name of the dashboard to be produced. Example: "my-freyja-dashboard" | Required | |
| freyja_dashboard | freyja_demixed | Array[File] | An array containing the output files (freyja_demixed) made by Freyja_FASTQ workflow | Required | |
| freyja_dashboard | samplename | Array[String] | The names of the samples being analyzed | Required | |
| freyja_dashboard | viral_load | Array[String] | An array containing the number of viral copies per liter | Required | |
| freyja_dashboard_task | config | File | (found in the optional section, but is required) A yaml file that applies various configurations to the dashboard, such as grouping lineages together, applying colorings, etc. See also https://github.com/andersen-lab/Freyja/blob/main/freyja/data/plot_config.yml. | Optional, Required | |
| freyja_dashboard | dashboard_intro_text | File | A file containing the text to be contained at the top of the dashboard. | SARS-CoV-2 lineage de-convolution performed by the Freyja workflow (https://github.com/andersen-lab/Freyja). | Optional |
| freyja_dashboard_task | cpu | Int | Number of CPUs to allocate to the task | 1 | Optional |
| freyja_dashboard_task | disk_size | Int | Amount of storage (in GB) to allocate to the task | 100 | Optional |
| freyja_dashboard_task | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/staphb/freyja:2.0.1 | Optional |
| freyja_dashboard_task | headerColor | String | A hex color code to change the color of the header | Optional | |
| freyja_dashboard_task | memory | Int | Amount of memory/RAM (in GB) to allocate to the task | 2 | Optional |
| freyja_dashboard_task | mincov | Float | The minimum genome coverage used as a cut-off of data to include in the dashboard. Default is set to 60 by the freyja command-line tool (not a WDL task default, per se) | Optional | |
| freyja_dashboard_task | scale_by_viral_load | Boolean | If set to true, averages samples taken the same day while taking viral load into account | False | Optional |
| freyja_dashboard_task | thresh | Float | The minimum lineage abundance cut-off value | Optional | |
| version_capture | docker | String | The Docker container to use for the task | us-docker.pkg.dev/general-theiagen/theiagen/alpine-plus-bash:3.20.0 | Optional |
| version_capture | timezone | String | Set the time zone to get an accurate date of analysis (uses UTC by default) | Optional |
Analysis Tasks¶
freyja_dashboard_task Details
This task will aggregate multiple samples together, and then create an interactive HTML visualization. Several optional inputs dictate the dashboard appearance (see each variable's description for more information).
Freyja Dashboard Technical Details
| Links | |
|---|---|
| Task | task_freyja_dashboard.wdl |
| Software Source Code | https://github.com/andersen-lab/Freyja |
| Software Documentation | https://github.com/andersen-lab/Freyja |
Outputs¶
| Variable | Type | Description |
|---|---|---|
| freyja_dashboard | File | The HTML file of the dashboard created |
| freyja_dashboard_metadata | File | The metadata used to create the dashboard |
| freyja_dashboard_version | String | The version of Freyja used |
| freyja_dashboard_wf_analysis_date | String | The date of analysis |
| freyja_dashboard_wf_version | String | The version of the Public Health Bioinformatics (PHB) repository used |
| freyja_demixed_aggregate | File | A TSV file that summarizes the freyja_demixed outputs for all samples |
Running Freyja on other pathogens¶
Experimental Feature
Please be aware this is an experimental feature and we cannot guarantee complete functionality at this time.
The main requirement to run Freyja on other pathogens is the existence of a barcode file for your pathogen of interest. Currently, barcodes exist for the following organisms:
- SARS-CoV-2 (default)
- FLU-B-VIC
- H1N1
- H3N2
- H5Nx-cattle
- H5NX
- MEASLESN450
- MEASLESgenome
- MPX
- RSVa
- RSVb
Freyja barcodes for other pathogens
Data for various pathogens can be found in the following repository: Freyja Barcodes
Folders are organized by pathogen, with each subfolder named after the date the barcode was generated, using the format YYYY-MM-DD, as well as a "latest" folder. Barcode files are named barcode.csv, and reference genome files are named reference.fasta.
There are two ways to run Freyja_FASTQ_PHB for non-SARS-CoV-2 organisms:
- Using the
freyja_pathogenoptional input (limited set of allowable organisms) - Providing the appropriate barcode file through the
freyja_barcodesoptional input (any organism for which barcodes are supplied)
Using the freyja_pathogen flag¶
When using the freyja_pathogen flag, the user must set the optional update_db flag to true, so that the latest version of the barcode file is automatically downloaded by Freyja.
Figure 2: Optional input for Freyja_FASTQ_PHB to provide the pathogen to be used by Freyja
Figure 2¶

Allowed options:
- SARS-CoV-2 (default)
- MPXV
- H1N1pdm
- H5NX
- FLU-B-VIC
- MEASLESN450
- MEASLES
- RSVa
- RSVb
Warning
The freyja_pathogen flag is not used if a barcodes file is provided. This means that this option is ignored if a barcode file is provided through freyja_barcodes.
Providing the appropriate barcode file¶
The appropriate barcode file for your organism of interest and reference sequence need to be downloaded and uploaded to your Terra.bio workspace. When running Freyja_FASTQ_PHB, the appropriate reference and barcodes file need to be passed as inputs. The first is a required input and will show up at the top of the workflows inputs page on Terra.bio (Figure 3).
Figure 3: Required input for Freyja_FASTQ_PHB to provide the reference genome to be used by Freyja
Figure 3¶

The barcodes file can be passed directly to Freyja by the freyja_barcodes optional input (Figure 4).
Figure 4: Optional input for Freyja_FASTQ_PHB to provide the barcodes file to be used by Freyja
Figure 4¶

References¶
If you use any of the Freyja workflows, please cite:
Karthikeyan, S., Levy, J.I., De Hoff, P. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108 (2022). https://doi.org/10.1038/s41586-022-05049-6
Freyja source code can be found at https://github.com/andersen-lab/Freyja
Freyja barcodes (non-SARS-CoV-2): https://github.com/gp201/Freyja-barcodes